Forme autosomique récessive connue syndrome de délétion multiple de l'ADNmt- l'encéphalomyopathie neurogastro-intestinale dite mitochondriale (MNGIE). La maladie se manifeste à un âge plus jeune et se manifeste par le même ensemble de symptômes neurologiques typiques des maladies mitochondriales en association avec un dysfonctionnement sévère du tractus gastro-intestinal (syndrome de pseudo-obstruction intestinale avec crises de vomissements répétés, diarrhée, amaigrissement).
Chez les malades MNGIE une diminution prononcée de l'activité de l'enzyme thymidine phosphorylase a été révélée, en raison de mutations du gène correspondant sur le chromosome 22ql3.32-qter. Ainsi, la maladie est basée sur une pathologie génétiquement déterminée du métabolisme de la thymidine, conduisant à une réplication et/ou à un maintien altérés de la molécule d'ADNmt.
syndrome de déplétion de l'ADNmt est une condition dans laquelle les patients n'ont pas un défaut qualitatif, mais un défaut quantitatif de l'ADNmt - c'est-à-dire une forte diminution du nombre de copies de molécules d'ADNmt (Fig. 68 C). De manière caractéristique, la déplétion de l'ADNmt n'est observée que dans des tissus strictement définis (par exemple, uniquement dans les muscles, uniquement dans le foie, les muscles et les reins, etc.).
Image clinique dépend de l'implication de tissus spécifiques et comprend généralement une myopathie (y compris congénitale), un syndrome convulsif, une insuffisance hépatique et rénale, une cardiomyopathie dans diverses combinaisons. Caractérisé par l'acidose lactique, le phénomène de "fibres rouges déchirées", a trouvé une insuffisance combinée de complexes de la chaîne respiratoire contenant des sous-unités codées par l'ADNmt (I, III-V).
Maladie est congénitale ou se manifeste au cours de la 1ère ou de la 2ème année de vie et est généralement mortelle. Dans la majorité des cas décrits de syndrome de déplétion de l'ADNmt, un type de transmission autosomique récessif a été enregistré. Le défaut génétique n'a pas été identifié. On suppose que la maladie est causée par des dommages au gène nucléaire qui contrôle la réplication de l'ADNmt. Une forme secondaire relativement bénigne de ce syndrome est également connue, dans laquelle la déplétion de l'ADNmt dans les cellules est causée par l'utilisation du médicament anti-VIH zidovudine.
Types de pathologie de l'ADN mitochondrial. Le site responsable du développement de l'épilepsie myoclonique à fibres rouges déchirées MERRF est présentéEn raison des fonctionnalités la génétique encéphalomyopathies mitochondriales Le diagnostic DQ de ces maladies présente un certain nombre de différences fondamentales par rapport aux approches traditionnelles applicables aux maladies mendéliennes. La généralisation de nombreuses années d'expérience dans l'examen de patients atteints de pathologie mitochondriale dans les principaux centres de recherche du monde a permis de développer un algorithme de diagnostic clair et cohérent qui fournit la recherche de diagnostic la plus efficace [Krasnopolskaya KD, Zakharova ELO., 1998; Shoffner J., Wallace D., 1992; DiMauro S., 1993; Chinnery P. et al., 1999 (a)]. Cet algorithme comprend plusieurs étapes principales.
A la première étape, il s'agit clinique et généalogique détaillée et l'analyse instrumentale de laboratoire, dont le but est d'accumuler des preuves en faveur d'une hypothèse éclairée sur la nature mitochondriale de la maladie à l'étude. Les signes les plus évidents de maladie mitochondriale sont :
a) type d'hérédité maternelle (en tenant compte de toutes les manifestations polymorphes et même subcliniques chez les frères et sœurs - enfants d'une mère malade);
b) la nature particulière du syndrome (présence d'une pathologie multisystémique et multiorganique impliquant des organes qui diffèrent par leur origine embryonnaire et leurs fonctions) ;
c) évolution progressive, présence de crises métaboliques (cette dernière est particulièrement importante pour les syndromes mitochondriaux de la petite enfance);
d) une augmentation du taux de lactate dans le sang et le liquide céphalo-rachidien (y compris dans le contexte de l'alimentation et de l'activité physique);
e) aminoacidurie et acidurie organique.
Recherche de pathologie multi-organes doit être effectuée de manière ciblée, en utilisant les méthodes paracliniques nécessaires, afin de détecter une cardiomyopathie manifeste ou cachée, une tubulopathie rénale, un dysfonctionnement hépatocellulaire, un diabète, un déficit en hormone de croissance, une atrophie des villosités intestinales, des modifications du frottis sanguin et des ponctuations médullaires, une myopathie, une , neuropathie cochléaire et visuelle, pathologie de la rétine, pétrifications et modifications focales de la substance cérébrale.
message mis à jour le 28/02/2019introduction(caractéristiques des mitochondries humaines). Une caractéristique du fonctionnement des mitochondries est la présence de leur propre génome mitochondrial - l'ADN mitochondrial circulaire (ADNmt) contenant 37 gènes, dont les produits sont impliqués dans le processus de production d'énergie dans la chaîne respiratoire des mitochondries. L'ADNmt est situé dans la membrane interne des mitochondries et se compose de cinq complexes enzymatiques conjugués, qui ont un total de 86 sous-unités. Ils sont principalement codés par des gènes nucléaires (ADNn), mais sept sous-unités du premier complexe enzymatique (ND1, 2, 3, 4, 4L, 5, 6), une du troisième (cytochrome b), trois du quatrième (COI , COII, COIII) et deux des cinquièmes (ATPase 6 et 8) sont codés par des gènes de structure d'ADNmt. Ainsi, des complexes enzymatiques (c'est-à-dire des protéines) codés à la fois par des gènes nucléaires (ADNn) et mitochondriaux (ADNmt) sont impliqués dans la garantie des diverses fonctions biochimiques des mitochondries.
Remarque! Les principaux processus biochimiques liés au métabolisme énergétique et se produisant dans les mitochondries sont : le cycle de l'acide tricarboxylique (cycle de Krebs), la bêta-oxydation des acides gras, le cycle de la carnitine, le transport d'électrons dans la chaîne respiratoire et la phosphorylation oxydative. Chacun de ces processus peut être perturbé et provoquer une insuffisance mitochondriale.
Cause de la maladie mitochondriale (ci-après MB). Les principales propriétés du génome mitochondrial sont l'héritage cytoplasmique des gènes, l'absence de recombinaisons (c'est-à-dire la réorganisation du matériel génétique par l'échange de segments individuels, de régions, de doubles hélices d'ADN) et un taux de mutation élevé. Le génome mitochondrial se caractérise par une instabilité prononcée et un taux élevé de substitutions de nucléotides, en moyenne 10 à 17 fois supérieur au taux de mutation des gènes nucléaires, et des mutations somatiques s'y produisent souvent au cours de la vie d'un individu. La cause immédiate de l'apparition et du développement du dysfonctionnement mitochondrial réside dans les défauts du système de phosphorylation oxydative, l'imperfection des mécanismes de réparation, l'absence d'histones et la présence de radicaux libres d'oxygène, qui sont des sous-produits de la respiration aérobie.
Les mutations du génome mitochondrial sont caractérisées par le phénomène [ !!! ] hétéroplasmie, dans laquelle (en raison de la spécificité de l'héritage mitochondrial), à la suite de la division cellulaire, la distribution (qui varie considérablement - de 1 à 99%) de l'ADNmt mutant entre les cellules filles se produit de manière aléatoire et inégale, à la suite de quelles copies d'ADNmt portant l'allèle normal et/ou mutant. Dans le même temps, différents tissus du corps ou des zones voisines du même tissu peuvent différer par le degré d'hétéroplasmie, c'est-à-dire selon le degré de présence et le rapport dans les cellules du corps des mitochondries avec de l'ADNmt mutant et normal (dans les générations suivantes, certaines cellules peuvent n'avoir que de l'ADNmt normal, une autre partie uniquement mutante et une troisième partie - les deux types d'ADNmt) . Le contenu des mitochondries avec l'ADNmt mutant augmente progressivement. En raison de cette "période de latence" (de l'anglais "lag" - delay), les futurs patients atteignent souvent la maturité sexuelle (et donnent une progéniture, portant presque toujours les mêmes mutations dans l'ADNmt). Lorsque le nombre de copies mutantes d'ADNmt dans une cellule atteint un certain seuil de concentration, le métabolisme énergétique dans les cellules est significativement altéré et se manifeste sous la forme d'une maladie (remarque : une caractéristique de MB héréditaire est souvent l'absence totale de tout élément pathologique signes au début de la vie du patient).
Remarque! L'hétéroplasmie est caractérisée par l'existence simultanée d'ADNmt mutant et normal dans la même cellule, tissu ou organe, ce qui détermine la gravité, la nature et l'âge de la manifestation MB. Le nombre d'ADNmt altéré peut également augmenter avec l'âge sous l'influence de divers facteurs et atteindre progressivement un niveau pouvant provoquer des manifestations cliniques de la maladie.
Conformément aux caractéristiques ci-dessus du génome mitochondrial double, le type d'héritage MB peut être différent. Étant donné que l'ADNmt dans le corps est presque exclusivement d'origine maternelle, lorsqu'une mutation mitochondriale est transmise à la progéniture, un type d'héritage maternel se produit dans le pedigree - tous les enfants d'une mère malade tombent malades. Si une mutation survient dans un gène nucléaire (ADNn) codant pour la synthèse d'une protéine mitochondriale, la maladie se transmet selon les lois mendéliennes classiques. Parfois, une mutation de l'ADNmt (généralement une délétion) survient de novo à un stade précoce de l'ontogenèse, puis la maladie se manifeste comme un cas sporadique.
Remarque! Actuellement, plus de 100 mutations ponctuelles et plusieurs centaines de réarrangements structurels de l'ADNmt sont connus pour être associés à des syndromes neuromusculaires et mitochondriaux caractéristiques allant de létaux pendant la période néonatale de la vie à des maladies d'apparition tardive.
Définition. MB peut être caractérisé comme des maladies causées par des défauts génétiques et biochimiques structurels des mitochondries et accompagnées d'une violation de la respiration tissulaire et, par conséquent, d'un défaut systémique du métabolisme énergétique, à la suite duquel les tissus et cibles les plus dépendants de l'énergie les organes sont touchés selon diverses combinaisons : cerveau, muscles squelettiques et myocarde (encéphalomyopathies mitochondriales), pancréas, organe de la vision, reins, foie. Cliniquement, les violations de ces organes peuvent être réalisées à tout âge. Parallèlement, l'hétérogénéité des symptômes complique le diagnostic clinique de ces maladies. La nécessité d'exclure MB se pose en présence de manifestations multisystémiques qui ne rentrent pas dans le processus pathologique habituel. La fréquence du dysfonctionnement de la chaîne respiratoire est estimée de 1 pour 5 à 10 000 à 4 à 5 pour 100 000 nouveau-nés.
Sémiotique. La pathologie neuromusculaire dans MB est généralement représentée par la démence, les convulsions, l'ataxie, la neuropathie optique, la rétinopathie, la surdité neurosensorielle, la neuropathie périphérique et la myopathie. Cependant, environ 1/3 des patients MB ont une intelligence normale et aucune manifestation neuromusculaire. MB comprend notamment l'encéphalocardiomyopathie de Kearns-Sayre (rétinite pigmentaire, ophtalmoplégie externe, bloc cardiaque complet) ; Syndrome MERRF (épilepsie myoclonique, fibres rouges « déchirées ») ; (encéphalomyopathie mitochondriale, acidose lactique, épisodes de type AVC) ; Syndrome de Pearson (encéphalomyopathie, ataxie, démence, ophtalmoplégie externe progressive) ; syndrome NAPR (neuropathie, ataxie, rétinite pigmentaire); et certaines formes de myopathie ophtalmopathique. Toutes ces formes sont réunies par un syndrome myopathique exprimé à un degré ou à un autre.
Remarque! Les deux principaux signes cliniques de MB sont l'augmentation au fil du temps du nombre d'organes et de tissus impliqués dans le processus pathologique, ainsi que les dommages presque inévitables au système nerveux central. Le polymorphisme des manifestations cliniques, y compris les lésions organiques, à première vue, physiologiquement et morphologiquement sans rapport, combiné à différentes périodes de manifestation et à la progression régulière des symptômes de la maladie avec l'âge, permet de suspecter une mutation [génétique] de l'ADNmt.
Remarque! Dans la pratique clinique, la capacité à différencier le tableau clinique du MB des conditions somatiques, auto-immunes, endocriniennes et autres pathologies plus courantes, dont la plupart sont traitables, est d'une grande importance. Il est nécessaire de procéder à une évaluation approfondie des antécédents familiaux, des données des méthodes d'examen cliniques et de laboratoire instrumentales de routine, avant d'attribuer des tests génétiques et biochimiques spécifiques au patient, visant à rechercher une pathologie mitochondriale.
Diagnostique . L'algorithme de diagnostic de tout MB doit inclure les étapes suivantes : [ 1 ] identification d'un tableau clinique typique du syndrome mitochondrial ou d'une lésion multisystémique "inexplicable" et d'une histoire héréditaire confirmant le type d'hérédité maternelle ; [ 2 ] une recherche diagnostique plus approfondie doit viser à détecter des marqueurs communs de dysfonctionnement mitochondrial : une augmentation du taux de lactate/pyruvate dans le sérum sanguin et le liquide céphalo-rachidien, une violation du métabolisme des glucides, des protéines et des acides aminés, ainsi qu'un tableau clinique impliquant au moins trois de ces systèmes dans le processus pathologique : système nerveux central, système cardiovasculaire, musculaire, endocrinien, rénal, organes de la vision et de l'ouïe ; [ 3 ] en cas de signes biologiques et instrumentaux cliniques et confirmés de pathologie mitochondriale, une analyse PCR des lymphocytes sanguins est réalisée pour la recherche ciblée de mutations ponctuelles de l'ADNmt ; une étude qui est considérée comme l'étalon-or pour diagnostiquer MB [cytopathies] - une biopsie des muscles squelettiques avec des analyses génétiques histochimiques, microscopiques électroniques, immunologiques et moléculaires, dont les changements caractéristiques seront avec n'importe quel MB (voir ci-dessous); [ 5 ] les tests les plus sensibles pour le diagnostic de MB sont les méthodes d'évaluation du niveau d'hétéroplasmie pathologique de l'ADNmt dans divers organes et tissus : PCR fluorescente, clonage, chromatographie liquide haute performance dénaturante, séquençage, hybridation Southern blot, etc.
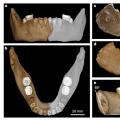
L'étude histochimique d'échantillons de biopsie musculaire de patients, y compris la coloration au trichrome selon la méthode de Gomori, démontre des changements caractéristiques de MB - fibres rouges déchirées de myofibrilles, qui contiennent un grand nombre de mitochondries proliférantes et endommagées, formant des agglomérats le long de la périphérie de la fibre musculaire . Dans ce cas, le nombre de fibres rouges déchirées dans la biopsie doit être ≥ 2 %. L'analyse enzymatique-histochimique montre un déficit en cytochrome C-oxydase dans 2 et 5% des myofibrilles (pour les patients de moins de 50 ans et de plus de 50 ans) de leur nombre total dans les échantillons de biopsie. L'analyse histochimique de l'activité de la succinate déshydrogénase (SDH) démontre une coloration positive à la CDH des myofibrilles (fibres bleues en lambeaux), qui, en combinaison avec une coloration positive à la SDH des parois artérielles qui irriguent les muscles, indique un degré élevé de dommages aux mitochondries des myocytes. Lors de la microscopie électronique d'échantillons de biopsie musculaire, des inclusions pathologiques, des réarrangements structurels des mitochondries, des modifications de leur forme, de leur taille et de leur nombre sont déterminés.
Remarque! Malgré des progrès significatifs depuis la découverte des mutations génétiques de l'ADNmt, la plupart des méthodes de diagnostic utilisées en pratique clinique ont un faible degré de spécificité pour les MB individuels. Par conséquent, les critères de diagnostic pour un MB particulier consistent tout d'abord en une combinaison de schémas cliniques et morphologiques spécifiques.
Principes de traitement . Le traitement du MB (cytopathies) est exclusivement symptomatique et vise à réduire le taux de progression de la maladie, ainsi qu'à améliorer la qualité de vie des patients. À cette fin, on prescrit aux patients une combinaison standard de médicaments, y compris la coenzyme Q10, l'idébénone - un analogue synthétique de la CoQ10, la créatine, l'acide folique, les vitamines B2, B6, B12 et d'autres médicaments qui améliorent les réactions redox dans les cellules (médicaments porteurs d'électrons dans la chaîne respiratoire et les cofacteurs des réactions enzymatiques du métabolisme énergétique). Ces composés stimulent la synthèse des molécules d'ATP et réduisent l'activité des processus radicalaires dans les mitochondries. Pendant ce temps, selon une revue systématique, la plupart des médicaments ayant des effets antioxydants et métaboliques utilisés dans MB n'ont pas été évalués dans de grands essais randomisés contrôlés par placebo. Par conséquent, il est difficile d'évaluer la sévérité de leur effet thérapeutique et la présence d'effets secondaires importants.
En savoir plus sur MB dans les sources suivantes:
 article "Pathologie neuromusculaire dans les maladies mitochondriales" L.A. Saykova, V.G. Pustozers; Académie médicale de formation postdoctorale de Saint-Pétersbourg de Roszdrav (magazine "Bulletin de l'Académie médicale de formation postdoctorale de Saint-Pétersbourg" 2009) [lire];
article "Pathologie neuromusculaire dans les maladies mitochondriales" L.A. Saykova, V.G. Pustozers; Académie médicale de formation postdoctorale de Saint-Pétersbourg de Roszdrav (magazine "Bulletin de l'Académie médicale de formation postdoctorale de Saint-Pétersbourg" 2009) [lire];
article "Maladies chroniques de genèse non inflammatoire et mutations du génome mitochondrial humain" K.Yu. Mitrofanov, A.V. Zhelankin, M.A. Sazonova, I.A. Sobenin, A.Yu. Postnov ; Centre d'innovation de Skolkovo. Institut de recherche sur l'athérosclérose, Moscou ; Institut de recherche GBOU de pathologie générale et de physiopathologie de l'Académie russe des sciences médicales, Moscou ; Institut de cardiologie clinique. A.L. Myasnikova FGBU RKNPK du ministère de la Santé et du Développement social de la Fédération de Russie (magazine "Cardiology Bulletin" n° 1, 2012) [lire] ;
article "ADN mitochondrial et pathologie héréditaire humaine" N.S. Prokhorov, L.A. Demidenko ; Département de biologie médicale, institution d'État "Université médicale d'État de Crimée nommée d'après I.I. SI. Georgievsky", Simferopol (magazine "Tauride Medical and Biological Bulletin" n° 4, 2010) [lire] ;
article "Génome mitochondrial et maladies mitochondriales humaines" I.O. Mazunin, N.V. Volodko, E.B. Starikovskaya, R.I. Sukernik ; Institut de biologie chimique et de médecine fondamentale, branche sibérienne de l'Académie russe des sciences, Novossibirsk (revue "Biologie moléculaire" n° 5, 2010) [lire] ;
article "Perspectives pour la médecine mitochondriale" par D.B. Zorov, N.K. Isaev, E.Yu. Plotnikov, D.N. Silachev, L.D. Zorova, I.B. Pevzner, MA Morosanova, S. S. Yankauskas, SD Zorov, V.A. Babenko ; Université d'Etat de Moscou M.V. Lomonosov, Institut de biologie physique et chimique nommé d'après A.I. UNE. Belozersky, Institut de recherche en mitoingénierie, Centre de recherche laser, Faculté de bioingénierie et de bioinformatique ; Université nationale russe de médecine de recherche. NI Pirogov (magazine "Biochimie" n° 9, 2013) [lire] ;
article "AVC dans les maladies mitochondriales" N.V. Pizov ; Département des maladies nerveuses avec des cours de neurochirurgie et de génétique médicale, SBEI HPE "Académie médicale d'État de Yaroslavl" (revue "Neurologie, neuropsychiatrie, psychosomatique" n° 2, 2012) [lire] ;
article "Diagnostic et prévention des maladies mitochondriales à codage nucléaire chez les enfants" E.A. Nikolaïev ; Institut clinique de recherche en pédiatrie, Moscou (Bulletin russe de périnatalogie et de pédiatrie, n° 2, 2014) [lire] ;
article "Épilepsie chez les enfants atteints de maladies mitochondriales : caractéristiques du diagnostic et du traitement" Zavadenko N.N., Kholin A.A. ; GBOU VPO Université médicale de recherche nationale russe. NI Pirogov du ministère de la Santé et du Développement social de Russie, Moscou (revue "Épilepsie et conditions paroxystiques" n° 2, 2012) [lire] ;
article "Pathologie mitochondriale et problèmes de la pathogenèse des troubles mentaux" par V.S. Soukhoroukov ; Institut de recherche de Moscou en pédiatrie et chirurgie pédiatrique de Rosmedtekhnologii (Journal of Neurology and Psychiatry, No. 6, 2008) [lire] ;
article "Algorithme pour le diagnostic des encéphalomyopathies mitochondriales" S.N. Illarioshkin (Nervous Diseases magazine No. 3, 2007) [lire];
article "Problèmes actuels de traitement des troubles mitochondriaux" par V.S. Soukhoroukov ; Institution budgétaire de l'État fédéral "Institut de recherche de Moscou sur la pédiatrie et la chirurgie pédiatrique" du ministère de la Santé de Russie (revue "Pharmacothérapie efficace. Pédiatrie" n° 4, 2012 [lire] ;
article "Leucoencéphalopathie avec lésion prédominante du tronc cérébral, de la moelle épinière et augmentation du lactate en spectroscopie IRM (observation clinique)" V.I. Guzeva, E.A. Efet, O.M. Nikolaeva; Université de médecine pédiatrique de Saint-Pétersbourg, Saint-Pétersbourg, Russie (revue « Neurochirurgie et neurologie de l'enfance » n° 1, 2013) [lire] ;
aide pédagogique pour les étudiants de troisième année de la faculté de diagnostic médical des universités de médecine "Maladies mitochondriales héréditaires" T.S. Ugolnik, I.V. Manaenkova; Établissement d'enseignement "Gomel State Medical University", Département de physiologie pathologique, 2012 [lire];
vite: Maladies mitochondriales(neurodégénérescence) - vers le site avec 17 liens vers des sources (articles, présentations, etc.).
© Laesus De Liro
Les maladies mitochondriales sont un groupe hétérogène de maladies causées par des dommages à certaines structures des cellules humaines qui sont essentielles à la conversion des aliments en énergie. Les maladies mitochondriales entraînent une diminution de la production d'énergie et des symptômes associés.
Les cellules sont les éléments constitutifs du corps humain, ce sont des structures microscopiques qui sont associées à une membrane et contiennent de nombreux composants - les organites, responsables de fonctions telles que la reproduction cellulaire, le transport de matériaux et la synthèse des protéines. La respiration cellulaire, le processus par lequel les molécules alimentaires sont converties en molécules à haute énergie utilisées comme source d'énergie, se déroule dans des structures appelées mitochondries. L'énergie mitochondriale est essentielle pour toutes les fonctions cellulaires.
On ne savait pas grand-chose sur les maladies mitochondriales jusqu'au milieu du XXe siècle. Le premier diagnostic d'un trouble mitochondrial a été posé en 1959 et le matériel génétique de l'ADNmt a été découvert en 1963. Dans les années 70 et 80 du siècle dernier, on en savait beaucoup plus sur les mitochondries, et le groupe des troubles mitochondriaux s'élargit à ce jour. La recherche dans les années 1990 a conduit à la classification des maladies mitochondriales.
Troubles mitochondriaux courants
À ce jour, il existe plus de quarante maladies mitochondriales différentes. Certains des troubles les plus courants comprennent:
Syndrome de Kearns-Sayre (KSS). Le KSS survient généralement avant l'âge de 20 ans. Les symptômes comprennent une difficulté progressive des mouvements oculaires, des paupières tombantes, une faiblesse musculaire, une petite taille, une perte auditive, une perte de coordination, des problèmes cardiaques, des retards cognitifs et le diabète.
 Épilepsie myocloniqueà fibres rouges cassées (MERRF). Le MERFF est un syndrome mitochondrial dans lequel un défaut mitochondrial ainsi qu'une anomalie tissulaire appelée "fibres rouges déchirées" sont détectés au microscope. Les symptômes comprennent des convulsions, une perte de coordination, une petite taille, une accumulation d'acide lactique dans le sang, des difficultés à parler, une démence et une faiblesse musculaire.
Épilepsie myocloniqueà fibres rouges cassées (MERRF). Le MERFF est un syndrome mitochondrial dans lequel un défaut mitochondrial ainsi qu'une anomalie tissulaire appelée "fibres rouges déchirées" sont détectés au microscope. Les symptômes comprennent des convulsions, une perte de coordination, une petite taille, une accumulation d'acide lactique dans le sang, des difficultés à parler, une démence et une faiblesse musculaire.
Encéphalomyopathie mitochondriale avec acidose lactique et accident vasculaire cérébral (MELAS). MELAS est une maladie évolutive, le syndrome mitochondrial affecte plusieurs systèmes d'organes, y compris le système nerveux central, le muscle cardiaque, le muscle squelettique et le tractus gastro-intestinal. Les symptômes comprennent une faiblesse musculaire, un accident vasculaire cérébral, une paralysie des muscles oculaires et une déficience cognitive.
Neuropathie optique héréditaire de Leber(LHON). LHON provoque une perte progressive de la vision conduisant à divers degrés de cécité et affecte principalement les hommes de plus de 20 ans. Des anomalies cardiaques peuvent également survenir.
Syndrome de Lee. Cette maladie cérébrale dégénérative est généralement diagnostiquée à un jeune âge. La détérioration s'accompagne souvent de symptômes tels que convulsions, démence, difficultés d'alimentation et d'élocution, dysfonctionnement respiratoire, problèmes cardiaques et faiblesse musculaire. Le pronostic est généralement sombre et la mort survient en quelques années.
Encéphalomyopathie neurogastro-intestinale mitochondriale (MNGIE). Les principaux signes incluent des symptômes qui imitent une obstruction gastro-intestinale et des anomalies du système nerveux. D'autres symptômes peuvent inclure une paralysie des muscles oculaires, une faiblesse musculaire, une perte de coordination et des anomalies cérébrales.
Syndrome de Person. Les premiers symptômes apparaissent généralement dans l'enfance, les caractéristiques de ce syndrome rare étant mises en évidence par un dysfonctionnement pancréatique et une anémie. Complications - obésité, diarrhée, hypertrophie du foie et autres signes.
Neuropathie, ataxie et rétinite pigmentaire (NARP). Les symptômes de ce trouble comprennent des troubles du système nerveux, une perte de coordination et une perte progressive de la vision. Peut également entraîner des retards de développement, de la démence, une faiblesse musculaire. Se produit généralement dans l'enfance.
Causes des troubles mitochondriaux
Bien que les maladies mitochondriales puissent être causées par des dommages au matériel génétique mitochondrial et affecter ainsi l'une des centaines de réactions chimiques nécessaires pour convertir l'oxygène et les nutriments en énergie, elles ont toutes un point commun : la capacité des mitochondries à produire de l'énergie est altérée. Les déchets de nombreuses réactions peuvent commencer à s'accumuler dans les cellules et interférer avec d'autres réactions chimiques, et au fil du temps, endommager davantage les mitochondries.
Hérédité des maladies mitochondriales
 Dans de nombreux cas, un trouble mitochondrial est transmis génétiquement de parent à enfant. Cela peut souvent être utile pour déterminer le type d'héritage. Les défauts génétiques peuvent être transmis par l'ADNn, le matériel génétique qui détermine la plupart des caractéristiques héréditaires, ou par l'ADNmt. Certains types de troubles mitochondriaux héréditaires comprennent :
Dans de nombreux cas, un trouble mitochondrial est transmis génétiquement de parent à enfant. Cela peut souvent être utile pour déterminer le type d'héritage. Les défauts génétiques peuvent être transmis par l'ADNn, le matériel génétique qui détermine la plupart des caractéristiques héréditaires, ou par l'ADNmt. Certains types de troubles mitochondriaux héréditaires comprennent :
Transmission autosomique récessive. Chaque personne possède deux ensembles de gènes, chacun hérité d'un parent. Dans le cas de certaines maladies génétiques, une personne doit avoir deux copies du gène défectueux pour avoir des symptômes de la maladie ; et si un seul des deux gènes est défectueux, alors la personne est considérée comme porteuse. Dans la transmission autosomique récessive, un individu reçoit le gène défectueux de chaque parent.
héritage maternel. L'ADNmt n'est transmis de la mère à l'enfant que parce que les mitochondries du spermatozoïde se trouvent dans la queue du spermatozoïde, qui n'est pas impliquée dans la conception. Certains troubles mitochondriaux ne peuvent donc être transmis que de la mère à l'enfant.
Hérédité récessive du chromosome X. Le sexe d'un enfant est déterminé par l'hérédité de brins d'ADN appelés chromosomes. Une fille hérite de deux chromosomes X, tandis qu'un garçon hérite d'un chromosome X d'un parent et d'un chromosome Y de l'autre. Si le gène défectueux codant pour la maladie se trouve sur le chromosome X, alors l'enfant mâle ne peut pas avoir une copie saine du gène (car il n'a qu'un seul chromosome X) ; ainsi, il aura des troubles. Les filles sont moins à risque car elles doivent avoir deux copies du gène défectueux (une sur chaque chromosome X) pour développer la maladie.
 Transmission autosomique dominante. Contrairement à la transmission autosomique récessive, une seule copie défectueuse du gène doit être héritée pour que la maladie se développe, de sorte que l'enfant a 50% de chances de développer la maladie.
Transmission autosomique dominante. Contrairement à la transmission autosomique récessive, une seule copie défectueuse du gène doit être héritée pour que la maladie se développe, de sorte que l'enfant a 50% de chances de développer la maladie.
Dans certains cas, des personnes sans facteur génétique souffrent du syndrome mitochondrial. Ces cas sont dits occasionnels ou sporadiques et peuvent être causés par diverses causes, notamment certains médicaments (tels que ceux utilisés pour traiter le VIH), l'anorexie, l'exposition à certaines toxines, des périodes prolongées de privation d'oxygène ou l'âge des parents.
Symptômes du syndrome mitochondrial
Étant donné que plus de 90 % de l'énergie nécessaire au corps humain est générée par les mitochondries, les effets des troubles mitochondriaux peuvent être considérables. La recherche montre que le cerveau, les nerfs, les muscles squelettiques, le foie, le cœur, les reins, les prothèses auditives, les yeux et le pancréas sont particulièrement touchés en raison des besoins énergétiques élevés. Certains des symptômes les plus courants de la maladie mitochondriale dans les systèmes d'organes sont les suivants :

D'autres symptômes comprennent des troubles du développement chez les jeunes enfants, une croissance médiocre, une petite taille, une fatigue accrue, des difficultés à respirer, à avaler et un risque accru d'infections.
Diagnostic des maladies mitochondriales
 L'éventail des symptômes qui se manifestent chez les enfants souffrant de troubles mitochondriaux est commun à de nombreuses autres maladies. Souvent, une caractéristique d'un trouble mitochondrial qui le distingue d'autres maladies présentant des symptômes similaires est des symptômes supplémentaires qui ne sont généralement pas présents dans une maladie non mitochondriale.
L'éventail des symptômes qui se manifestent chez les enfants souffrant de troubles mitochondriaux est commun à de nombreuses autres maladies. Souvent, une caractéristique d'un trouble mitochondrial qui le distingue d'autres maladies présentant des symptômes similaires est des symptômes supplémentaires qui ne sont généralement pas présents dans une maladie non mitochondriale.
En raison de la nature complexe des troubles mitochondriaux, les médecins adoptent des approches à multiples facettes pour diagnostiquer ces maladies. Le processus commence généralement par un examen médical complet, une évaluation des antécédents médicaux et familiaux du patient. Souvent, un examen neurologique est effectué pour déterminer s'il existe des anomalies cérébrales. Des tests plus approfondis peuvent être effectués pour diagnostiquer le syndrome mitochondrial et exclure d'autres maladies. Certaines de ces méthodes de test sont les suivantes :
évaluation initiale. La première ligne de tests comprend généralement les méthodes les moins invasives, telles que le test d'un échantillon de sang pour évaluation. Dans certains cas, le diagnostic peut être posé sur la base de tests sanguins; dans d'autres, des tests sanguins peuvent indiquer que des tests supplémentaires sont nécessaires.
Évaluation secondaire. Ces tests peuvent être plus intenses, plus agressifs et/ou comporter plus de risques. Les exemples incluent la ponction lombaire, l'analyse d'urine, l'imagerie par résonance magnétique (IRM), les tests sanguins supplémentaires, l'électrocardiogramme (ECG).
Évaluation tertiaire. Procédures compliquées et/ou invasives telles que les tests cutanés ou la biopsie musculaire. Dans certains cas, des tests tertiaires sont nécessaires pour établir un diagnostic définitif.
Dans certaines situations, un médecin peut ne pas être en mesure de diagnostiquer un patient atteint d'un trouble mitochondrial particulier, même après une évaluation minutieuse. Par conséquent, il convient de garder à l'esprit que, malgré la difficulté de tester les troubles mitochondriaux, leur diagnostic n'est pas toujours possible.
Traitement des maladies mitochondriales
Il n'existe pas de médicaments spécifiques pour le traitement des troubles mitochondriaux. Le plan de traitement vise principalement à retarder la progression de la maladie ou à réduire les symptômes du patient. Les méthodes de traitement dépendent de nombreux facteurs, notamment du type de maladie, de l'âge de la personne, des organes touchés et de son état de santé. Tous les patients ne bénéficient pas du traitement.
 La thérapie, quant à elle, peut consister en des cures de vitamines, de compléments nutritionnels, de kinésithérapie ou d'ergothérapie, de médecines traditionnelles, telles que :
La thérapie, quant à elle, peut consister en des cures de vitamines, de compléments nutritionnels, de kinésithérapie ou d'ergothérapie, de médecines traditionnelles, telles que :
- des vitamines telles que les vitamines B (thiamine, riboflavine, niacine, acide folique, biotine et acide pantothénique), la vitamine E, la vitamine C,
- la coenzyme Q10 (CoQ10), impliquée dans la respiration cellulaire dans les mitochondries normales,
- la lévocarnitine, prise par voie orale ou administrée par voie intraveineuse,
- thérapie antioxydante,
- physiothérapie ou ergothérapie pour les myopathies.
Pour certains patients, la minimisation des facteurs physiologiques tels que le froid extrême, les températures élevées, une mauvaise alimentation, le jeûne et le manque de sommeil peut améliorer leur état. L'alcool, la fumée de cigarette et le glutamate monosodique peuvent également exacerber les troubles mitochondriaux.
Dans certains cas, un régime alimentaire bien conçu est nécessaire pour éviter l'aggravation des symptômes. Les parents d'un enfant atteint du syndrome mitochondrial doivent consulter un nutritionniste pour créer un régime alimentaire individualisé. Un régime alimentaire individualisé peut inclure de petits repas fréquents, augmenter ou diminuer l'apport en matières grasses et éviter ou compléter certaines vitamines ou minéraux.
Nouvelle recherche
Les scientifiques recherchent des médicaments pour traiter les maladies mitochondriales. Le problème est compliqué par le fait que ces maladies sont très rares: par exemple, le nombre total de patients atteints de MELAS ne dépasse pas 60 000 personnes dans le monde, ce qui rend non rentable le développement de médicaments pour ces maladies. Malgré cela, des médicaments sont néanmoins apparus assez efficaces pour lutter contre les manifestations de la pathologie mitochondriale.
Ainsi, pour le traitement de l'ataxie de Friedreich, on utilise le médicament EPI-743, qui a montré son efficacité dans plusieurs études. Cet outil vous permet d'optimiser la production d'énergie dans les mitochondries et de réduire le déséquilibre redox.
Dans le traitement de l'encéphalomyélopathie (MELAS), un certain effet positif a été montré par la L-arginine, dont l'administration intraveineuse et orale a permis de réduire la sévérité des principaux symptômes de cette maladie : céphalées, nausées avec vomissements, troubles visuels et conscience. Cela a été démontré dans une étude de 9 ans menée par des scientifiques japonais.
Pronostic des troubles mitochondriaux
Le pronostic d'une maladie mitochondriale individuelle dépend de nombreux facteurs, y compris le trouble spécifique, le mode de transmission, l'âge du patient et les organes affectés. Par exemple, deux enfants souffrant de la même maladie mitochondriale peuvent suivre deux traitements complètement différents. Dans certains cas, les patients peuvent être en mesure de contrôler leurs symptômes dans une large mesure avec différentes procédures ou si la progression de la maladie est lente. Dans d'autres cas, la maladie progresse rapidement et conduit à la mort inévitable.
Dans le cas d'un enfant à risque de troubles mitochondriaux, les parents peuvent être intéressés par un conseil génétique. Les tests génétiques, cependant, ne peuvent pas déterminer avec précision comment et quand un enfant peut développer une maladie mitochondriale ou sa gravité.
Déni de responsabilité : Les informations fournies dans cet article sur les maladies mitochondriales sont destinées à informer le lecteur uniquement. Il ne peut se substituer à l'avis d'un professionnel de santé.
Les maladies mitochondriales sont un groupe de pathologies héréditaires résultant de troubles de l'énergie cellulaire, caractérisées par un polymorphisme des manifestations cliniques, exprimées dans la lésion prédominante du système nerveux central et du système musculaire, ainsi que d'autres organes et systèmes du corps.
Une définition alternative de la pathologie mitochondriale dit qu'il s'agit d'un grand groupe de conditions pathologiques causées par des défauts génétiques, structurels et biochimiques des mitochondries, une altération de la respiration des tissus et, par conséquent, un métabolisme énergétique insuffisant.
Comme le souligne A. Munnich, "les maladies mitochondriales peuvent provoquer n'importe quel symptôme, dans n'importe quel tissu, à n'importe quel âge, avec n'importe quel type d'hérédité".
Les chaînes respiratoires mitochondriales sont la principale voie finale du métabolisme aérobie. Par conséquent, la pathologie mitochondriale est souvent appelée "maladies de la chaîne respiratoire mitochondriale" (MRDC); Il s'agit d'une classe de maladies relativement nouvelle.
Aspects historiques de la pathologie mitochondriale
R. Luft et al. (1962) ont trouvé une relation entre la faiblesse musculaire et les perturbations des processus de phosphorylation oxydative dans le tissu musculaire. S. Nass et M. Nass (1963) ont découvert l'existence de leur propre appareil génétique de mitochondries (plusieurs copies du chromosome en anneau ont été trouvées). En 1960-1970. le concept de maladies mitochondriales est apparu, c'est-à-dire une pathologie médiée étiologiquement par un dysfonctionnement mitochondrial. Dans les années 1980 des preuves génétiques moléculaires précises de la nature mitochondriale d'un certain nombre de maladies (maladie de Leber, syndrome de Pearson) ont été obtenues.
Aspects étiopathogéniques de la pathologie mitochondriale
Selon la présence du principal défaut métabolique, il est d'usage de considérer quatre grands groupes de maladies mitochondriales : 1) les troubles du métabolisme du pyruvate ; 2) anomalies du métabolisme des acides gras ; 3) violations du cycle de Krebs ; 4) défauts de transport d'électrons et de phosphorylation oxydative (OXPHOS).
Les causes de la pathologie mitochondriale sont des mutations dans les gènes codant pour les protéines impliquées dans le métabolisme énergétique des cellules (y compris les sous-unités du complexe pyruvate déshydrogénase, les enzymes du cycle de Krebs, les composants de la chaîne de transport d'électrons, les protéines structurelles de la chaîne de transport d'électrons (ETC), les cellules internes mitochondriales). transporteurs membranaires, régulateurs du pool de nucléotides mitochondriaux, ainsi que des facteurs interagissant avec l'ADN mitochondrial (ADNmt).
Les troubles mitochondriaux sont associés à un grand nombre de maladies qui ne sont pas des cytopathies mitochondriales primaires. Néanmoins, dans ces maladies, les dysfonctionnements mitochondriaux contribuent de manière significative à la pathogenèse et aux manifestations cliniques des maladies. Les maladies décrites peuvent être des malformations métaboliques, dégénératives, inflammatoires, congénitales/acquises et des néoplasmes.
La mitochondrie est un organite présent dans presque toutes les cellules, à l'exception des globules rouges matures. C'est pourquoi les maladies mitochondriales peuvent affecter tous les systèmes et organes du corps humain. À cet égard, il est plus correct d'appeler ces conditions "cytopathies mitochondriales".
Les principales caractéristiques des cytopathies mitochondriales comprennent un polymorphisme prononcé des symptômes cliniques, une nature multisystémique de la lésion, une variabilité de l'évolution, une progression et une réponse inadéquate à la thérapie utilisée.
La chaîne respiratoire est localisée sur la membrane mitochondriale interne et comprend cinq complexes multienzymatiques, dont chacun, à son tour, se compose de plusieurs dizaines de sous-unités. L'ADN mitochondrial ne code que pour 13 des sous-unités protéiques de la chaîne respiratoire, 2 sous-unités protéiques de l'ARNmt et 22 ARN de transfert mitochondrial (ARNt). Le génome nucléaire code pour plus de 90 % des protéines mitochondriales.
Le résultat final de la phosphorylation oxydative se produisant dans les complexes 1-γ est la production d'énergie (ATP). L'adénosine triphosphate est la principale source d'énergie des cellules.
L'ADN mitochondrial interagit étroitement avec l'ADN nucléaire (ADNn). Dans chacun des 5 complexes respiratoires, la plupart des sous-unités sont codées par l'ADNn, et non par l'ADNmt. Le complexe I se compose de 41 sous-unités, dont 7 sont codées par l'ADNmt et le reste par l'ADNn. Le complexe II n'a que 4 sous-unités; la plupart d'entre eux sont codés par l'ADN nucléaire. Le complexe III est représenté par dix sous-unités ; codage de l'ADNmt - 1, ADNn - 9. Le complexe IV a 13 sous-unités, dont 3 sont codées par l'ADNmt et 10 par l'ADNn. Le complexe V comprend 12 sous-unités, l'ADNmt codant - 2, l'ADNn - 10.
Les violations de l'énergie cellulaire conduisent à des maladies polysystémiques. Tout d'abord, les organes et tissus les plus dépendants énergétiquement souffrent : le système nerveux (encéphalopathie, polyneuropathie), le système musculaire (myopathies), le cœur (cardiomyopathies), les reins, le foie, le système endocrinien et d'autres organes et systèmes. Jusqu'à récemment, toutes ces maladies étaient définies sous de nombreux masques d'autres formes nosologiques de pathologie. À ce jour, plus de 200 maladies causées par des mutations de l'ADN mitochondrial ont été identifiées.
Les maladies mitochondriales peuvent être causées par une pathologie des génomes mitochondrial et nucléaire. Comme l'ont souligné P. F. Chinnery et al. (2004) et S. DiMauro (2004), des mutations de l'ADNmt ont été détectées dans 1 cas pour 8 000 habitants et la prévalence des maladies mitochondriales est d'environ 11,5 cas pour 100 000 habitants.
Chaque cellule contient de plusieurs centaines à plusieurs milliers d'organites - mitochondries, contenant de 2 à 10 molécules circulaires d'ADN mitochondrial, capables de réplication, de transcription et de traduction, et indépendamment de l'ADN nucléaire.
Aspects génétiques de la pathologie mitochondriale
La génétique mitochondriale diffère de la génétique mendélienne classique par trois aspects importants : 1) l'hérédité maternelle (le cytoplasme entier, avec les organites qu'il contient, est reçu par la progéniture avec l'œuf) ; 2) hétéroplasmie - l'existence simultanée dans la cellule de types d'ADN normaux (sauvages) et mutants; 3) ségrégation mitotique (les deux types d'ADNmt dans le processus de division cellulaire peuvent être distribués de manière aléatoire entre les cellules filles).
L'ADN mitochondrial accumule les mutations plus de 10 fois plus vite que le génome nucléaire, car il manque d'histones protectrices et son environnement est extrêmement riche en espèces réactives de l'oxygène, qui sont un sous-produit des processus métaboliques se produisant dans les mitochondries. La proportion d'ADNmt mutant doit dépasser un seuil critique avant que les cellules ne commencent à présenter des anomalies biochimiques des chaînes respiratoires mitochondriales (effet de seuil). Le niveau de pourcentage d'ADNmt mutant peut varier entre les individus au sein des familles, ainsi que dans les organes et les tissus. C'est une des explications de la variabilité du tableau clinique chez les patients présentant des dysfonctionnements mitochondriaux. Les mêmes mutations peuvent provoquer différents syndromes cliniques (par exemple, mutation A3243G - encéphalopathie avec paroxysmes ressemblant à des accidents vasculaires cérébraux - syndrome MELAS, ainsi qu'ophtalmoplégie externe progressive chronique, diabète sucré). Des mutations dans différents gènes peuvent provoquer le même syndrome. L'exemple classique d'une telle situation est le syndrome MELAS.
Variétés de pathologie mitochondriale
Si nous énumérons les principales maladies mitochondriales, elles comprendront les suivantes : l'encéphalopathie neurogastro-intestinale mitochondriale (MNGIE), le syndrome de délétion multiple de l'ADN mitochondrial, la myopathie lipidique avec des niveaux normaux de carnitine, le déficit en carnitine palmitoyl transférase, le diabète sucré mitochondrial, la maladie d'Alpers-Huttenlocher, Syndrome de Kearns-Sayre, maladie de Leber (LHON), syndrome de Wolfram, syndrome MEMSA, syndrome de Pearson, syndrome SANDO, syndrome MIRAS, syndrome MELAS, syndrome MERRF, syndrome SCAE, syndrome NARP, syndrome de Barth, syndrome CPEO, syndrome de Lee, etc.
Les syndromes cliniques les plus fréquents de la pathologie mitochondriale chez l'enfant sont : le syndrome MELAS (encéphalomyopathie mitochondriale, acidose lactique et paroxysmes ressemblant à des accidents vasculaires cérébraux), le syndrome MERRF (épilepsie myoclonique avec fibres rouges déchiquetées), le syndrome de Kearns-Sayre (caractérisé par un ptosis, une ophtalmoplégie, une rétinite pigmentosa, ataxie, troubles de la conduction cardiaque), syndrome NARP (neuropathie, ataxie, rétinite pigmentaire), syndrome de Lee (encéphalomyélopathie nécrosante subaiguë), maladie de Leber (neuropathie optique héréditaire).
Il existe un grand nombre de maladies causées non pas par des mutations de l'ADN mitochondrial, mais par des mutations de l'ADN nucléaire qui codent pour la fonction mitochondriale. Il s'agit notamment des types de pathologie suivants : maladie de Barth (myopathie, cardiomyopathie, neutro- et thrombocytopénie transitoires), encéphalopathie gastro-intestinale mitochondriale (maladie multisystémique autosomique récessive) : ptosis, ophtalmoplégie, neuropathie périphérique, dysfonctionnement gastro-intestinal entraînant une cachexie, leucoencéphalopathie. L'âge de survenue de cette dernière maladie est très variable, allant de la période néonatale à 43 ans.
Diagnostic de la pathologie mitochondriale
Les critères cliniques de diagnostic des maladies mitochondriales sont relativement nombreux : 1) complexe de symptômes myopathiques (intolérance à l'exercice, faiblesse musculaire, diminution du tonus musculaire) ; 2) convulsions (myocloniques ou multifocales); 3) syndrome cérébelleux (ataxie, tremblement intentionnel); 4) lésions des nerfs oculomoteurs (ptosis, ophtalmoplégie externe) ; 5) polyneuropathie ; 6) paroxysmes ressemblant à des accidents vasculaires cérébraux ; 7) maux de tête de type migraine ; 8) dysmorphie craniofaciale ; 9) manifestations dysmétaboliques (vomissements, épisodes de léthargie, coma) ; 10) troubles respiratoires (apnée, hyperventilation, tachypnée) ; 11) dommages au cœur, au foie, aux reins ; 12) évolution progressive de la maladie.
Les critères cliniques suivants sont utilisés dans le diagnostic des maladies mitochondriales : 1) signes de lésions du tissu conjonctif (syndrome d'hypermobilité, hyperélasticité cutanée, troubles de la posture, etc.) ; 2) manifestations neurodégénératives, leucopathie au cours de l'imagerie par résonance magnétique (IRM) du cerveau ; 3) des épisodes répétés d'altération de la conscience ou des épisodes inexpliqués de vomissements chez les nouveau-nés ; 4) ataxie inexpliquée ; 5) retard mental sans raisons spécifiques ; 6) antécédents familiaux chargés; 7) une détérioration soudaine de l'état de l'enfant (convulsions, vomissements, troubles respiratoires, léthargie, faiblesse, altération du tonus musculaire - plus souvent hypotension musculaire, coma, léthargie; lésions hépatiques et rénales qui ne se prêtent pas au traitement conventionnel).
Les études de laboratoire (biochimiques) visent principalement à identifier l'acidose lactique et / ou l'acidose au pyruvate chez les patients. Il convient de rappeler que des taux normaux d'acide lactique n'excluent pas la présence d'une maladie mitochondriale. D'autres paramètres biochimiques étudiés en cas de suspicion de pathologie mitochondriale comprennent les corps cétoniques sanguins et urinaires, les acylcarnitines plasmatiques et les acides organiques et acides aminés sanguins et urinaires.
M.V. Miles et al. (2008) ont proposé d'évaluer le contenu en coenzyme musculaire Q10 chez les enfants présentant un défaut des enzymes de la chaîne respiratoire mitochondriale.
Des études cytomorphodensitométriques permettent d'évaluer l'activité des mitochondries lymphocytaires (diminution du nombre, augmentation du volume, diminution de l'activité).
A partir d'études instrumentales (en plus des méthodes de neuroimagerie), une biopsie du muscle squelettique est utilisée avec des réactions histochimiques spécifiques pour identifier le phénomène de "fibres rouges déchiquetées" (ragged red fibres - RRF) dans la biopsie résultante. Les syndromes à "fibres rouges déchirées" sont les suivants : MELAS, MERRF, KSS, PEO (ophtalmoplégie externe progressive) et syndrome de Pearson. Syndromes sans RRF : maladie de Leigh, NARP, LHON (neuropathie optique héréditaire de Leber).
Les méthodes de recherche génétique se réduisent à la détermination des mutations les plus fréquentes et au séquençage de l'ADN mitochondrial.
Traitement de la pathologie mitochondriale
La thérapie pour les maladies mitochondriales, malheureusement, n'a pas été développée. Du point de vue de la médecine factuelle, on pense qu'il n'existe pas de traitement efficace pour ce groupe représentatif de maladies. Néanmoins, dans divers pays du monde, des agents pharmacologiques et des substances biologiquement actives sont utilisés pour normaliser le métabolisme et fournir une énergie adéquate aux mitochondries.
Dans le syndrome MELAS, le traitement doit viser à traiter les convulsions, les troubles endocriniens et à éliminer les conséquences d'un accident vasculaire cérébral.
P. Kaufmann et al. (2006) indiquent que puisque les niveaux de lactate sont souvent en corrélation avec la sévérité des manifestations neurologiques, il est raisonnable d'utiliser le dichloroacétate pour réduire les niveaux de lactate. Dans notre pays, le diméthyloxobutylphosphonyl diméthylate (Dimephosphone) est utilisé dans un but similaire.
Dans les études des auteurs japonais Y. Koga et al. (2002, 2005, 2006, 2007), l'administration intraveineuse de L-arginine (précurseur du NO) a été utilisée avec un bon effet pour stimuler la vasodilatation dans la période aiguë de l'AVC, ainsi que l'administration orale pour réduire la gravité des épisodes ultérieurs.
Parmi les médicaments utilisés dans le traitement de la pathologie mitochondriale figurent les suivants: vitamine B 1 (thiamine) - 400 mg / jour, vitamine B 2 (riboflavine) - 100 mg / jour, vitamine C (acide ascorbique) - jusqu'à 1 g / jour, vitamine E (tocophérol) - 400 UI / jour, nicotinamide (niacine) - jusqu'à 500 mg / jour, coenzyme Q 10 - de 90 à 200 mg / jour, L-carnitine - de 10 mg à 1-2 g / jour, acide succinique - de 25 mg à 1,5 g / jour, Dimephosphone 15% - 1,0 ml pour 5 kg de poids corporel. Le cytochrome C (intraveineux), la Reamberin (intraveineuse) et la cytoflavine (intraveineuse et orale) sont également utilisés.
Les autres moyens de pharmacothérapie sont les corticostéroïdes, les minéralocorticoïdes (avec le développement d'une insuffisance surrénalienne), les anticonvulsivants - avec convulsions / épilepsie (à l'exclusion de l'acide valproïque et de ses dérivés, limitant l'utilisation des barbituriques). Dans nos observations, le traitement anticonvulsivant le plus efficace était l'utilisation du lévétiracétam (Keppra), du topiramate (Topamax) ou de leurs combinaisons.
Neurodiétologie en pathologie mitochondriale
Le principe principal du régime alimentaire en pathologie mitochondriale est la restriction des nutriments qui ont un effet négatif sur les mécanismes métaboliques - jusqu'à la formation d'un bloc métabolique (le régime est simultanément enrichi d'autres composants à un niveau normal ou élevé). Cette stratégie thérapeutique a été appelée « faire le tour du bloc ». Une exception importante à cet égard est le groupe des troubles mitochondriaux associés au métabolisme du pyruvate (insuffisance du complexe pyruvate déshydrogénase avec troubles concomitants glucides/glycogène/acides aminés). Cependant, le régime cétogène et d'autres types de régimes riches en graisses sont recommandés.
Les substances qui sont des cofacteurs alimentaires sont largement utilisées (coenzyme Q 10, L-carnitine, acétyl-L-carnitine, vitamine B 2, acide ascorbique, vitamine E, vitamine B 1, nicotinamide, vitamine B 6, vitamine B 12, biotine, folique , vitamine K, acide α-lipoïque, acide succinique, Se) . Il est recommandé d'éviter les facteurs nutritionnels individuels qui induisent une exacerbation de la maladie mitochondriale (famine, consommation de graisses, protéines, saccharose, amidon, alcool, caféine, glutamate monosodique ; troubles alimentaires quantitatifs et apport insuffisant d'énergie alimentaire). Si nécessaire, une nutrition clinique est assurée (entérale, parentérale, gastrostomie).
Le diagnostic opportun des maladies mitochondriales, la recherche de critères cliniques et paracliniques pour ces maladies au stade préliminaire prégénétique sont extrêmement importants. Cela est nécessaire pour sélectionner une thérapie métabolique adéquate et prévenir la détérioration ou l'invalidité chez les patients atteints de ces maladies rares.
C. S. Chi (2015) souligne que la confirmation ou l'exclusion de la pathologie mitochondriale reste fondamentale en pratique pédiatrique, notamment lorsque les signes cliniques de la maladie ne sont pas spécifiques, ce qui nécessite une approche de suivi pour évaluer les symptômes et les paramètres biochimiques.
Littérature
- Martikainen M.H., Chinnery P.F. Maladie mitochondriale : mimiques et caméléons // Pract. Neurol. 2015. Vol. 15(6):424-435.
- Sarnat H.B., Menkes J.H. Encéphalomyopathies mitochondriales. Ch. 2. Dans : Child Neuroloy (Menkes J. H., Sarnat H. B., Maria B. L., eds). 7e éd. Philadelphie-Baltimore. Lippincott Williams & Wilkins. 2006. 143-161.
- Luft R., Ikkos D., Palmieri G., Ernster L., Afzelius B. Un cas d'hypermétabolisme sévère d'origine non thyroïdienne avec un défaut de maintien du contrôle respiratoire mitochondrial : une étude clinique, biochimique et morphologique corrélée // J. Clin. Investir. 1962 Vol. 41 : 1776-1804.
- Nass M.M., Nass S. Fibres intramitochondriales avec des caractéristiques d'ADN. I. Réactions de fixation et de coloration des électrons // J. Cell. Biol. 1963 Vol. 19:593-611.
- Nass S., Nass M.M. Fibres intramitochondriales avec des caractéristiques d'ADN. II. Traitements enzymatiques et autres traitements hydrolytiques // J. Cell. Biol. 1963 Vol. 19:613-629.
- Sukhorukov V.S. Essais sur la pathologie mitochondriale. M. : Medpraktika-M, 2011. 288 p.
- Chinnery PF, DiMauro S., Shanske S., Schon EA, Zeviani M., Mariotti C., Carrara F., Lombes A., Laforet P., Ogier H., Jaksch M., Lochmuller H., Horvath R., Deschauer M., Thorburn DR, Bindoff LA, Poulton J., Taylor RW, Matthews JN, Turnbull DM Risque de développer un trouble de délétion de l'ADN mitochondrial // Lancet. 2004. 364 (9434): 592-596.
- DiMauro S. Maladies mitochondriales // Biochim. Biophys. acte. 2004. 1658(1-2) : 80-88.
- Siciliano G., Volpi L., Piazza S., Ricci G., Mancuso M., Murri L. Diagnostic fonctionnel dans les maladies mitochondriales // Biosci. représentant 2007 Vol. 27(1-3): 53-67.
- Miles M.V., Miles L., Tang P.H., Horn P.S., Steele P.E., DeGrauw A.J., Wong B.L., Bove K.E.Évaluation systématique de la teneur en coenzyme musculaire Q10 chez les enfants présentant des déficits en enzymes de la chaîne respiratoire mitochondriale // Mitochondrion. 2008 Vol. 8(2): 170-180.
- Kaufmann P., Engelstad K., Wei Y., Jhung S., Sano MC, Shungu DC, Millar WS, Hong X., Gooch CL, Mao X., Pascual JM, Hirano M., Stacpoole PW, DiMauro S., De Vivo DC Le dichloracétate provoque une neuropathie toxique dans MELAS: un essai clinique contrôlé randomisé // Neurology. 2006 Vol. 66(3): 324-330.
- Lignes directrices fédérales pour l'utilisation des médicaments (système de formulaire). Publier. XVI. M. : Ekho, 2015. 540.
- Koga Y., Ishibashi M., Ueki I., Yatsuga S., Fukiyama R., Akita Y., Matsuishi T. Effets de la L-arginine sur la phase aiguë des accidents vasculaires cérébraux chez trois patients atteints de MELAS // Neurology. 2002 Vol. 58(5): 827-828.
- Koga Y., Akita Y., Nishioka J., Yatsuga S., Povalko N., Tanabe Y., Fujimoto S., Matsuishi T. La L-arginine améliore les symptômes des épisodes ressemblant à des accidents vasculaires cérébraux dans MELAS // Neurology. 2005 Vol. 64(4): 710-712.
- Koga Y., Akita Y., Junko N., Yatsuga S., Povalko N., Fukiyama R., Ishii M., Matsuishi T. Dysfonction endothéliale dans MELAS améliorée par la supplémentation en L-arginine // Neurology. 2006 Vol. 66(11): 1766-1769.
- Koga Y., Akita Y., Nishioka J., Yatsuga S., Povalko N., Katayama K., Matsuishi T. Thérapie MELAS et L-arginine // Mitochondrie. 2007 Vol. 7(1-2): 133-139.
- Rai P.K., Russell O.M., Lightowlers R.N., Turnbull D.M. Composés potentiels pour le traitement de la maladie mitochondriale // Br. Méd. Taureau. 2015. 20 novembre. pii : ldv046. .
- Finsterer J., Bindu P.S. Stratégies thérapeutiques pour les troubles mitochondriaux // Pediatr. Neurol. 2015. Vol. 52(3): 302-313.
- Studenikin V.M., Goryunova A.V., Gribakin S.G., Zhurkova N.V., Zvonkova N.G., Ladodo K.S., Pak L.A., Roslavtseva E.A., Stepakina E.I., Studenikina N.I., Tursunkhuzhaeva S.Sh., Shelkovsky V.I. Encéphalopathies mitochondriales. Chapitre 37. Dans le livre : Neurodiétologie de l'enfance (monographie collective) / Ed. Studenikina V.M.M. : Dynasty, 2012. S. 415-424.
- Chi C. S. Approche diagnostique chez les nourrissons et les enfants atteints de maladies mitochondriales // Pediatr. néonatal. 2015. Vol. 56(1): 7-18.
V. M. Studenikin* , 1 ,docteur en sciences médicales, professeur, académicien de l'Académie russe des sciences naturelles
O.V. Globa**,Candidat en sciences médicales
* GOU VPO RNIMU eux. N. I. Pirogov Ministère de la Santé de la Fédération de Russie, Moscou
** GOU VPO PMGMU eux. I. M. Sechenov Ministère de la Santé de la Fédération de Russie, Moscou
Pathologie mitochondriale et problèmes de la pathogenèse des troubles mentaux
VS. Soukhoroukov
La pathologie mitochondriale et les problèmes de physiopathologie des troubles mentaux
VS. Soukhoroukov
Institut de recherche de Moscou en pédiatrie et chirurgie pédiatrique, Rosmedtekhnologii
Au cours des dernières décennies, une nouvelle direction s'est activement développée en médecine, associée à l'étude du rôle des troubles du métabolisme énergétique cellulaire - processus qui affectent les organites cellulaires universels - les mitochondries. À cet égard, le concept de "maladies mitochondriales" est apparu.
Les mitochondries remplissent de nombreuses fonctions, mais leur tâche principale est la formation de molécules d'ATP dans les cycles biochimiques de la respiration cellulaire. Les principaux processus se produisant dans les mitochondries sont le cycle des acides tricarboxyliques, l'oxydation des acides gras, le cycle de la carnitine, le transport d'électrons dans la chaîne respiratoire (à l'aide des complexes enzymatiques I-IV) et la phosphorylation oxydative (complexe enzymatique V). Les dysfonctionnements mitochondriaux sont parmi les stades les plus importants (souvent précoces) des dommages cellulaires. Ces troubles conduisent à un apport énergétique insuffisant aux cellules, à la perturbation de nombreux autres processus métaboliques importants, au développement ultérieur de dommages cellulaires pouvant aller jusqu'à la mort cellulaire. Pour le clinicien, l'évaluation du degré de dysfonctionnement mitochondrial est essentielle à la fois pour la formation d'idées sur la nature et l'étendue des processus se produisant au niveau tissulaire, et pour l'élaboration d'un plan de correction thérapeutique de l'état pathologique.
Le concept de "maladies mitochondriales" s'est formé en médecine à la fin du 20e siècle en raison de maladies héréditaires découvertes peu de temps auparavant, dont les principaux facteurs étiopathogéniques sont des mutations des gènes responsables de la synthèse des protéines mitochondriales. Tout d'abord, les maladies associées aux mutations de l'ADN mitochondrial découvertes au début des années 1960 ont été étudiées. Cet ADN, qui a une structure relativement simple et ressemble au chromosome circulaire des bactéries, a été étudié en détail. La structure primaire complète de l'ADN mitochondrial humain (mitDNA) a été publiée en 1981), et déjà à la fin des années 1980, le rôle prépondérant de ses mutations dans le développement d'un certain nombre de maladies héréditaires a été prouvé. Ces derniers comprennent l'atrophie optique héréditaire de Leber, le syndrome NARP (neuropathie, ataxie, rétinite pigmentaire), le syndrome MERRF (épilepsie myoclonique avec fibres rouges « déchirées » dans les muscles squelettiques), le syndrome MELAS (encéphalomyopathie mitochondriale, acidose lactique, épisodes de type accident vasculaire cérébral), Syndrome de Kearns-Sayre (rétinite pigmentaire, ophtalmoplégie externe, bloc cardiaque, ptosis, syndrome cérébelleux), syndrome de Pearson (atteinte médullaire, dysfonctionnement pancréatique et hépatique), etc. Le nombre de descriptions de ces maladies augmente chaque année. Selon les dernières données, la fréquence cumulée des maladies héréditaires associées aux mutations du mitDNA atteint 1/5000 personnes dans la population générale.
Dans une moindre mesure, les défauts mitochondriaux héréditaires associés à des dommages au génome nucléaire ont été étudiés. A ce jour, relativement peu d'entre eux sont connus (diverses formes de myopathies infantiles, maladies d'Alpers, de Ley, de Barth, de Menkes, syndromes de carence en carnitine, certaines enzymes du cycle de Krebs et de la chaîne respiratoire des mitochondries). On peut supposer que leur nombre devrait être beaucoup plus important, puisque les gènes codant l'information de 98 % des protéines mitochondriales sont situés dans le noyau.
En général, on peut dire que l'étude des maladies causées par des troubles héréditaires des fonctions mitochondriales a fait une sorte de révolution dans les idées modernes sur les aspects médicaux du métabolisme énergétique humain. Outre la contribution à la pathologie théorique et à la systématique médicale, l'une des principales réalisations de la "mitochondriologie" médicale a été la création d'une boîte à outils de diagnostic efficace (critères génétiques cliniques, biochimiques, morphologiques et moléculaires de l'insuffisance mitochondriale polysystémique), qui a permis pour évaluer les troubles polysystémiques du métabolisme énergétique cellulaire.
En ce qui concerne la psychiatrie, déjà dans les années 30 du XXe siècle, des données ont été obtenues selon lesquelles chez les patients atteints de schizophrénie, après l'exercice, le niveau d'acide lactique augmente fortement. Plus tard, sous la forme d'une hypothèse scientifique formalisée, le postulat est apparu que certains mécanismes de régulation des échanges énergétiques sont responsables du manque "d'énergie mentale" dans cette maladie. Cependant, pendant assez longtemps, de telles hypothèses ont été perçues comme, pour le moins, "peu prometteuses d'un point de vue scientifique". En 1965, S. Kety écrivait: "Il est difficile d'imaginer qu'un défaut généralisé du métabolisme énergétique - un processus fondamental pour chaque cellule du corps - puisse être responsable des caractéristiques hautement spécialisées de la schizophrénie". Cependant, la situation a changé au cours des 40 années suivantes. Les succès de la « médecine mitochondriale » étaient si convaincants qu'ils ont commencé à attirer l'attention d'un cercle plus large de médecins, y compris des psychiatres. Le résultat de la croissance constante du nombre d'études pertinentes a été résumé dans les travaux de A. Gardner et R. Boles "Does "mitochondrial psychiatry" have a future?" . La forme interrogative du postulat inclus dans le titre portait une nuance de modestie exagérée. Le volume d'informations fournies dans l'article était si important et la logique des auteurs était si parfaite qu'il n'y avait plus aucun doute sur les perspectives de la "psychiatrie mitochondriale".
À ce jour, il existe plusieurs groupes de preuves de l'implication de perturbations dans les processus énergétiques dans la pathogenèse de la maladie mentale. Chacun des groupes de preuves est discuté ci-dessous.
Troubles mentaux dans les maladies mitochondriales
Les différences dans le seuil de sensibilité des tissus à une production insuffisante d'ATP laissent une empreinte significative sur le tableau clinique des maladies mitochondriales. À cet égard, le tissu nerveux est principalement intéressant car il est le plus dépendant de l'énergie. De 40 à 60% de l'énergie de l'ATP dans les neurones est dépensée pour maintenir le gradient ionique sur leur enveloppe externe et la transmission de l'influx nerveux. Par conséquent, les dysfonctionnements du système nerveux central dans les "maladies mitochondriales" classiques sont d'une importance capitale et justifient d'appeler le principal complexe de symptômes "encéphalomyopathies mitochondriales". Dans le même temps, des troubles cérébraux tels que l'arriération mentale, les convulsions et les épisodes ressemblant à des accidents vasculaires cérébraux sont apparus cliniquement. La sévérité de ces pathologies associées à des troubles somatiques sévères peut être telle que d'autres troubles plus légers, associés notamment à des modifications de la personnalité ou des émotions, restent dans l'ombre.
L'accumulation d'informations sur les troubles mentaux dans les maladies mitochondriales a commencé à se produire beaucoup plus tard par rapport aux troubles ci-dessus. Néanmoins, il existe maintenant une quantité suffisante de preuves de leur existence. Des troubles affectifs dépressifs et bipolaires, des hallucinations et des changements de personnalité ont été décrits dans le syndrome de Kearns-Sayre, le syndrome MELAS, l'ophtalmoplégie externe progressive chronique et la neuropathie optique héréditaire de Leber.
Très souvent, le développement des signes classiques de la maladie mitochondriale est précédé de troubles mentaux modérément graves. Par conséquent, les patients peuvent être initialement observés par des psychiatres. Dans ces cas, d'autres symptômes de la maladie mitochondriale (photophobie, vertiges, fatigue, faiblesse musculaire, etc.) sont parfois considérés comme des troubles psychosomatiques. Le célèbre chercheur en pathologie mitochondriale P. Chinnery, dans un article écrit en collaboration avec D. Turnbull, souligne : « Des complications psychiatriques accompagnent constamment la maladie mitochondriale. Ils prennent généralement la forme d'une dépression réactive... Nous avons observé à plusieurs reprises des cas de dépression sévère et de tentatives de suicide avant même (soulignement ajouté par les auteurs de l'article) que le diagnostic ne soit établi.
Les difficultés à établir le véritable rôle des troubles mentaux dans les maladies considérées sont également liées au fait que les symptômes et syndromes psychiatriques peuvent être considérés dans certains cas comme une réaction à une situation difficile, dans d'autres comme la conséquence d'une lésion cérébrale organique ( dans ce dernier cas, le terme « psychiatrie » en général non utilisé).
Sur la base des matériaux d'un certain nombre de revues, nous présentons une liste de troubles mentaux décrits chez les patients atteints de formes avérées de maladies mitochondriales 1 . Ces violations peuvent être divisées en trois groupes. I. Troubles psychotiques - hallucinations (auditives et visuelles), symptômes de schizophrénie et d'états schizophréniques, délire. Dans certains cas, ces troubles font suite à une déficience cognitive progressive. II. Troubles affectifs et anxieux - états dépressifs bipolaires et unipolaires (ils sont décrits le plus souvent), états paniques, phobies. III. Trouble cognitif sous la forme d'un trouble déficitaire de l'attention avec hyperactivité. Ce syndrome a été décrit non seulement chez des patients diagnostiqués avec une maladie "mitochondriale", mais aussi chez leurs proches. En particulier, un cas est décrit lorsqu'une maladie basée sur la délétion d'une paire de nucléotides d'ADNmit dans la région du gène de l'ARN de transfert s'est manifestée pour la première fois au cours des années scolaires d'un garçon sous la forme d'un trouble déficitaire de l'attention avec hyperactivité. La progression de l'encéphalomyopathie mitochondriale a entraîné le décès de ce patient à l'âge de 23 ans. IV. Troubles de la personnalité. De tels troubles ont été décrits dans un certain nombre de cas avec des diagnostics confirmés par des études de génétique moléculaire. En règle générale, les troubles de la personnalité se développent après une déficience cognitive. Un cas d'autisme chez un patient présentant une mutation ponctuelle du mitDNA dans la région du gène de l'ARN de transfert est décrit.
Caractéristiques communes caractéristiques des maladies mitochondriales et psychiatriques
Nous parlons d'une certaine similitude clinique de certaines maladies mentales et syndromes mitochondriaux, ainsi que de types communs de leur transmission.
Tout d'abord, l'attention est attirée sur les données sur la prévalence des cas d'hérédité maternelle de certaines maladies mentales, en particulier les troubles bipolaires. Un tel héritage ne peut pas être expliqué en termes de mécanismes autosomiques, et le nombre égal d'hommes et de femmes parmi les patients atteints de troubles bipolaires rend peu probable qu'un héritage lié à l'X soit possible dans ce cas. L'explication la plus adéquate à cela pourrait être le concept de transmission d'informations héréditaires via le mitDNA. Il existe également une tendance à l'hérédité maternelle chez les patients atteints de schizophrénie. Certes, à cet égard, il existe une explication alternative utilisée dans notre contexte : on suppose que cette tendance peut être due à des conditions inégales pour les patients de sexes différents dans la recherche d'un partenaire.
La confirmation indirecte du lien entre les mitochondries et certaines maladies mentales est également une tendance à la cyclicité de leurs manifestations cliniques. Avec des maladies comme le trouble bipolaire, c'est de notoriété publique. Cependant, les données sur les rythmes ultra-, circadiens et saisonniers des manifestations cliniques des états dysénergétiques commencent également à s'accumuler en mitochondriologie. Cette caractéristique a même déterminé le nom de l'une de leurs cytopathies mitochondriales nosologiques - "syndrome des vomissements cycliques" ("syndrome des vomissements cycliques").
Enfin, la similitude envisagée des deux groupes de maladies apparaît dans les signes somatiques qui les accompagnent. Des symptômes psychosomatiques bien connus des psychiatres, tels que déficience auditive, douleurs musculaires, fatigue, migraines, syndrome du côlon irritable, sont constamment décrits dans le complexe symptomatique des maladies mitochondriales. Comme l'écrivent A. Gardner et R. Boles, "si le dysfonctionnement mitochondrial est l'un des facteurs de risque de développement de certaines maladies psychiatriques, ces symptômes somatiques comorbides peuvent être le résultat d'un dysfonctionnement mitochondrial plutôt qu'une manifestation de "détresse communicative", " schéma hypochondrial » ou « acquisition secondaire » (« gain secondaire ») ». Parfois, ces termes sont utilisés pour désigner le phénomène de somatisation des troubles mentaux.
En conclusion, nous soulignons une autre similitude : une augmentation de la densité de substance blanche déterminée par imagerie par résonance magnétique est notée non seulement dans les troubles affectifs bipolaires et la dépression majeure d'apparition tardive, mais également dans les cas de modifications ischémiques des encéphalopathies mitochondriales.
Signes de dysfonctionnement mitochondrial dans la maladie mentale
Schizophrénie
Comme mentionné ci-dessus, la mention de signes d'acidose lactique et de certains autres changements biochimiques, indiquant une violation du métabolisme énergétique dans la schizophrénie, a commencé à apparaître à partir des années 30 du XXe siècle. Mais ce n'est qu'à partir des années 1990 que le nombre de travaux pertinents a commencé à augmenter de manière particulièrement notable et que le niveau méthodologique de la recherche en laboratoire a également augmenté, ce qui s'est reflété dans un certain nombre de publications de synthèse.
Sur la base de travaux publiés, D. Ben-Shachar et D. Laifenfeld ont divisé tous les signes de troubles mitochondriaux dans la schizophrénie en trois groupes : 1) troubles morphologiques des mitochondries ; 2) signes d'une violation du système de phosphorylation oxydative; 3) des perturbations dans l'expression des gènes responsables des protéines mitochondriales. Cette division peut être étayée par des exemples tirés d'autres travaux.
L'autopsie du tissu cérébral de patients atteints de schizophrénie L. Kung et R. Roberts a révélé une diminution du nombre de mitochondries dans le cortex frontal, le noyau caudé et le putamen. Dans le même temps, il a été noté qu'il était moins prononcé chez les patients traités avec des antipsychotiques et, par conséquent, les auteurs ont estimé qu'il était possible de parler de la normalisation des processus mitochondriaux dans le cerveau sous l'influence d'un traitement antipsychotique. Cela donne lieu de mentionner l'article de N.S. Kolomeets et N.A. Uranova sur l'hyperplasie mitochondriale dans les terminaisons axonales présynaptiques dans le domaine de la substance noire dans la schizophrénie.
L. Cavelier et al. , examinant le matériel d'autopsie du cerveau de patients atteints de schizophrénie, a révélé une diminution de l'activité du complexe IV de la chaîne respiratoire dans le noyau caudé.
Ces résultats nous ont permis de suggérer un rôle primaire ou secondaire du dysfonctionnement mitochondrial dans la pathogenèse de la schizophrénie. Cependant, le matériel d'autopsie étudié était lié à des patients traités avec des antipsychotiques et, naturellement, des troubles mitochondriaux étaient associés à l'exposition aux médicaments. Notez que de telles hypothèses, souvent fondées, accompagnent toute l'histoire de la découverte des changements mitochondriaux dans divers organes et systèmes dans les maladies mentales et autres. En ce qui concerne l'influence possible des neuroleptiques eux-mêmes, il convient de rappeler que la tendance à l'acidose lactique chez les patients atteints de schizophrénie a été découverte dès 1932, près de 20 ans avant leur apparition.
Une diminution de l'activité de divers composants de la chaîne respiratoire a été constatée dans le cortex frontal et temporal, ainsi que dans les ganglions de la base du cerveau et d'autres éléments tissulaires - plaquettes et lymphocytes chez les patients atteints de schizophrénie. Cela a permis de parler du caractère polysystémique de l'insuffisance mitochondriale. S. Whatley et al. , en particulier, ont montré que dans le cortex frontal l'activité du complexe IV diminue, dans le cortex temporal - complexes I, III et IV; dans les ganglions de la base - complexes I et III, aucun changement n'a été trouvé dans le cervelet. Il convient de noter que l'activité de l'enzyme intramitochondriale, la citrate synthase, correspondait aux valeurs témoins dans toutes les zones étudiées, ce qui permettait de parler de la spécificité des résultats obtenus pour la schizophrénie.
Outre les études envisagées, on peut citer les travaux réalisés en 1999-2000. les travaux de J. Prince et al. qui ont étudié l'activité des complexes respiratoires dans différentes parties du cerveau de patients atteints de schizophrénie. Ces auteurs n'ont trouvé aucun signe de changement dans l'activité du complexe I, mais l'activité du complexe IV a été réduite dans le noyau caudé. Dans le même temps, cette dernière ainsi que l'activité du complexe II sont augmentées dans la coquille et dans le noyau accumbens. De plus, une augmentation de l'activité du complexe IV dans la coquille était significativement corrélée à la sévérité des dysfonctionnements émotionnels et cognitifs, mais pas au degré des troubles moteurs.
Il est à noter que les auteurs de la plupart des travaux cités ci-dessus attribuaient les signes de troubles du métabolisme énergétique aux effets des neuroleptiques. En 2002, des données très intéressantes à cet égard ont été publiées par A. Gardner et al. sur les enzymes mitochondriales et la production d'ATP dans des échantillons de biopsie musculaire de patients atteints de schizophrénie traités avec des antipsychotiques et non traités avec eux. Ils ont constaté qu'une diminution de l'activité des enzymes mitochondriales et de la production d'ATP a été constatée chez 6 patients sur 8 qui n'ont pas reçu d'antipsychotiques, et chez les patients sous traitement antipsychotique, une augmentation de la production d'ATP a été constatée. Ces données ont confirmé dans une certaine mesure les conclusions antérieures de L. Kung et R. Roberts.
En 2002, les résultats d'un autre travail remarquable ont été publiés. Il a étudié l'activité du complexe I de la chaîne respiratoire dans les plaquettes de 113 patients atteints de schizophrénie en comparaison avec 37 sains. Les patients ont été divisés en trois groupes: groupe 1 - avec un épisode psychotique aigu, groupe 2 - avec une forme active chronique et groupe 3 - avec une schizophrénie résiduelle. Les résultats ont montré que l'activité du complexe I était significativement augmentée par rapport au contrôle chez les patients des groupes 1 et 2 et diminuée chez les patients du groupe 3. De plus, une corrélation significative a été trouvée entre les paramètres biochimiques obtenus et la sévérité des symptômes cliniques de la maladie. Des changements similaires ont été obtenus dans l'étude des sous-unités flavoprotéiques du complexe I dans le même ARN et matériel protéique. Les résultats de cette étude ont donc non seulement confirmé la forte probabilité de défaillance mitochondriale multisystémique dans la schizophrénie, mais ont également permis aux auteurs de recommander des méthodes de laboratoire appropriées pour le suivi de la maladie.
Après 2 ans en 2004, D. Ben-Shachar et al. ont publié des données intéressantes sur l'effet de la dopamine sur la chaîne respiratoire des mitochondries, qui joue un rôle important dans la pathogenèse de la schizophrénie. Il a été découvert que la dopamine peut inhiber l'activité du complexe I et la production d'ATP. Dans le même temps, l'activité des complexes IV et V ne change pas. Il s'est avéré que, contrairement à la dopamine, la noradrénaline et la sérotonine n'affectent pas la production d'ATP.
Il convient de noter l'accent mis dans les travaux ci-dessus sur le dysfonctionnement du complexe I de la chaîne respiratoire mitochondriale. Ce type de changement peut refléter des perturbations relativement modérées de l'activité mitochondriale, qui sont plus importantes du point de vue de la régulation fonctionnelle du métabolisme énergétique que des baisses brutales (proche de la létalité pour la cellule) de l'activité de la cytochrome oxydase.
Arrêtons-nous maintenant brièvement sur l'aspect génétique de la pathologie mitochondriale dans la schizophrénie.
En 1995-1997 L. Cavelier et al. il a été constaté que le niveau de "délétion normale" du mitDNA (la délétion la plus courante de 4977 paires de bases, affectant les gènes des complexes des sous-unités I, IV et V et sous-jacente à plusieurs maladies mitochondriales graves, telles que le syndrome de Kearns-Sayre, etc. ) n'est pas modifié dans le matériel d'autopsie du cerveau de patients atteints de schizophrénie, ne s'accumule pas avec l'âge et n'est pas corrélé à une activité cytochrome oxydase altérée. En séquençant le génome mitochondrial chez des patients atteints de schizophrénie, les chercheurs de ce groupe ont montré la présence d'un polymorphisme du gène du cytochrome b différent du témoin.
Au cours de ces années, une série d'ouvrages du groupe R. Marchbanks et autres a également été publiée. qui ont étudié l'expression de l'ARN nucléaire et mitochondrial dans le cortex frontal dans des cas de schizophrénie. Ils ont découvert que toutes les séquences mises à l'échelle à partir du contrôle étaient liées à des gènes mitochondriaux. A été significativement augmenté, en particulier, l'expression du gène mitochondrial de la 2ème sous-unité de la cytochrome oxydase. Quatre autres gènes étaient liés à l'ARN ribosomal mitochondrial.
Des chercheurs japonais, examinant 300 cas de schizophrénie, n'ont pas trouvé de signes de la mutation 3243AG (qui provoque une violation du complexe I dans le syndrome MELAS). Aucune fréquence de mutation accrue n'a été trouvée dans les gènes mitochondriaux de la 2e sous-unité du complexe I, du cytochrome b et des ribosomes mitochondriaux dans la schizophrénie dans les travaux de K. Gentry et V. Nimgaonkar.
R. Marchbanks et al. ont trouvé une mutation dans la paire de nucléotides 12027 du mitDNA (gène de la 4ème sous-unité du complexe I), qui était présente chez les patients masculins atteints de schizophrénie et qui ne l'était pas chez les femmes.
La caractérisation de trois gènes nucléaires du complexe I a été étudiée dans le cortex préfrontal et visuel de patients atteints de schizophrénie par R. Karry et al. . Ils ont constaté que la transcription et la traduction de certaines sous-unités étaient réduites dans le cortex préfrontal et augmentées dans le cortex visuel (les auteurs ont interprété ces données conformément au concept d'"hypofrontalité" dans la schizophrénie). Dans l'étude des gènes (y compris les gènes des protéines mitochondriales) dans le tissu hippocampique des patients atteints de schizophrénie traités avec des antipsychotiques, aucun changement n'a été trouvé.
Les chercheurs japonais K. Iwamoto et al. , étudiant les modifications des gènes responsables de l'information héréditaire des protéines mitochondriales du cortex préfrontal dans la schizophrénie en lien avec un traitement neuroleptique, a obtenu des preuves en faveur d'effets médicamenteux sur le métabolisme énergétique cellulaire.
Les résultats ci-dessus peuvent être complétés par des données d'études intravitales, qui ont été examinées par W. Katon et al. : lors de l'étude de la distribution de l'isotope du phosphore 31P à l'aide de la spectroscopie par résonance magnétique, une diminution du niveau de synthèse d'ATP dans les ganglions de la base et le lobe temporal du cerveau de patients atteints de schizophrénie a été révélée.
Dépression et troubles affectifs bipolaires
Les chercheurs japonais T. Kato et al. la spectroscopie par résonance magnétique a révélé une diminution du pH intracellulaire et du niveau de phosphocréatine dans le lobe frontal du cerveau chez les patients atteints de troubles bipolaires, y compris ceux qui n'ont pas reçu de traitement. Les mêmes auteurs ont mis en évidence une diminution du taux de phosphocréatine dans le lobe temporal chez les patients résistants à la lithiumthérapie. D'autres auteurs ont trouvé une diminution des niveaux d'ATP dans le lobe frontal et les ganglions de la base de patients souffrant de dépression majeure. Notez que des signes similaires ont été observés chez des patients atteints de certaines maladies mitochondriales.
En ce qui concerne les données de génétique moléculaire, il convient de noter immédiatement que les résultats d'un certain nombre d'études indiquent l'absence de preuve de l'implication des délétions de l'ADNmit dans le développement des troubles de l'humeur.
Un certain nombre d'études sur le polymorphisme du mitDNA, outre le fait même de la différence de ses haplotypes chez les patients atteints de troubles bipolaires et examinés dans le groupe témoin, ont révélé certaines mutations caractéristiques du premier, en particulier aux positions 5178 et 10398 - les deux les positions sont dans la zone des gènes du complexe I.
Il existe des rapports sur la présence de mutations dans les gènes du complexe I, non seulement dans les mitochondries, mais aussi dans les gènes nucléaires. Ainsi, dans des cultures de cellules lymphoblastoïdes issues de patients atteints de troubles bipolaires, une mutation a été retrouvée dans le gène NDUFV2, localisé sur le 18e chromosome (18p11), et codant pour une des sous-unités du complexe I. Le séquençage MitDNA de patients atteints de troubles bipolaires a révélé une mutation caractéristique en position 3644 du gène de la sous-unité ND1, qui appartient également au complexe I. Une augmentation du niveau de traduction (mais pas de transcription) a été trouvée pour certaines sous-unités du complexe I dans le cortex visuel de patients atteints de trouble bipolaire. Parmi d'autres études, nous citerons deux études dans lesquelles les gènes de la chaîne respiratoire ont été investigués et leurs désordres génétiques moléculaires ont été retrouvés dans le cortex préfrontal et l'hippocampe de patients atteints de troubles bipolaires. Dans un des travaux de A. Gardner et al. chez les patients souffrant de dépression majeure, un certain nombre de troubles enzymatiques mitochondriaux et une diminution du niveau de production d'ATP dans les tissus musculo-squelettiques ont été détectés, et une corrélation significative a été trouvée entre le degré de diminution de la production d'ATP et les manifestations cliniques d'un trouble mental.
Autres troubles mentaux
Il existe peu de recherches sur le dysfonctionnement mitochondrial dans d'autres troubles psychiatriques. Certains d'entre eux ont été mentionnés dans les sections précédentes de l'examen. Ici, nous mentionnons spécifiquement les travaux de P. Filipek et al. , qui décrit 2 enfants autistes et une mutation du 15e chromosome, dans la région 15q11-q13. Les deux enfants présentaient un retard modéré du développement moteur, une léthargie, une hypotension sévère, une acidose lactique, une activité réduite du complexe III et une hyperprolifération mitochondriale dans les fibres musculaires. Ce travail est remarquable par le fait qu'il a été le premier à décrire des troubles mitochondriaux dans le complexe symptomatique d'une maladie associée étiologiquement à une région spécifique du génome.
Données généalogiques concernant le rôle possible des troubles mitochondriaux dans la pathogenèse de la maladie mentale
Ci-dessus, nous avons déjà mentionné une caractéristique d'un certain nombre de maladies mentales comme une fréquence accrue des cas d'hérédité maternelle, ce qui peut indiquer indirectement l'implication de la pathologie mitochondriale dans leur pathogenèse. Cependant, il existe des preuves plus convaincantes pour ce dernier dans la littérature.
En 2000, les données obtenues par F. McMahon et al sont publiées. qui a séquencé l'intégralité du génome mitochondrial chez 9 candidats non apparentés, tous issus d'une famille élargie avec transmission maternelle du trouble bipolaire. Il n'y avait pas de différences évidentes dans les haplotypes par rapport aux familles témoins. Cependant, pour certaines positions du mitDNA (709, 1888, 10398 et 10463) une disproportion entre les personnes malades et en bonne santé a été constatée. Dans le même temps, nous pouvons noter la coïncidence des données sur la position 10398 avec les données déjà mentionnées des auteurs japonais, qui ont suggéré que le polymorphisme 10398A mitDNA est un facteur de risque pour le développement de troubles bipolaires.
La preuve généalogique la plus significative du rôle des dysfonctionnements mitochondriaux dans le développement des troubles mentaux est le fait que les patients atteints de maladies mitochondriales classiques ont des parents (plus souvent du côté maternel) atteints de troubles mentaux modérés. L'anxiété et la dépression sont souvent mentionnées parmi ces troubles. Ainsi, dans les travaux de J. Shoffner et al. il a été constaté que la sévérité de la dépression chez les mères de patients "mitochondriaux" est 3 fois plus élevée que dans le groupe témoin.
Il convient de noter les travaux de B. Burnet et al. qui a mené une enquête anonyme auprès de patients atteints de maladies mitochondriales, ainsi que des membres de leur famille, pendant 12 mois. Parmi les questions étaient liées à l'état de santé des parents et des proches des patients (sur les lignées paternelle et maternelle). Ainsi, 55 familles (Groupe 1) avec une suspicion maternelle et 111 familles (Groupe 2) avec une suspicion de mode de transmission non maternelle de la maladie mitochondriale ont été étudiées. En conséquence, les parents des patients du côté maternel, par rapport au côté paternel, ont montré une incidence plus élevée de plusieurs conditions pathologiques. Parmi eux, avec les migraines et le syndrome du côlon irritable, il y avait la dépression. Dans le groupe 1, des dysfonctionnements intestinaux, des migraines et des dépressions ont été observés chez un plus grand pourcentage de mères des familles enquêtées - 60, 54 et 51 %, respectivement ; dans le 2ème groupe - dans 16, 26 et 12%, respectivement (p<0,0001 для всех трех симптомов). У отцов из обеих групп это число составляло примерно 9-16%. Достоверное преобладание указанных признаков имело место и у других родственников по материнской линии. Этот факт является существенным подтверждением гипотезы о возможной связи депрессии с неменделевским наследованием, в частности с дисфункцией митохондрий.
Aspects pharmacologiques de la pathologie mitochondriale dans la maladie mentale
Effet des médicaments utilisés en psychiatrie sur la fonction mitochondriale
Dans les sections précédentes de la revue, nous avons déjà brièvement abordé les questions thérapeutiques. En particulier, la question de l'effet possible des antipsychotiques sur les fonctions mitochondriales a été discutée. Il a été constaté que la chlorpromazine et d'autres dérivés de la phénothiazine, ainsi que les antidépresseurs tricycliques, peuvent affecter le métabolisme énergétique dans le tissu cérébral : ils peuvent réduire le niveau de phosphorylation oxydative dans certaines zones du cerveau, peuvent découpler l'oxydation et la phosphorylation, réduire l'activité de le complexe I et l'ATPase, abaissent le niveau d'utilisation de l'ATP. Cependant, l'interprétation des faits dans ce domaine requiert une grande prudence. Ainsi, le découplage de l'oxydation et de la phosphorylation sous l'influence des neuroleptiques n'a été nullement noté dans toutes les zones du cerveau (il n'est pas déterminé dans le cortex, le thalamus et le noyau caudé). De plus, il existe des données expérimentales sur la stimulation de la respiration mitochondriale par les neuroleptiques. Dans les sections précédentes de la revue, nous présentons également des travaux qui témoignent de l'effet positif des antipsychotiques sur la fonction mitochondriale.
La carbamazépine et le valproate sont connus pour leur capacité à supprimer la fonction mitochondriale. La carbamazépine entraîne une augmentation du taux de lactate dans le cerveau et le valproate est capable d'inhiber les processus de phosphorylation oxydative. Le même type d'effets (mais seulement à des doses élevées) a été révélé dans une étude expérimentale sur les inhibiteurs de la recapture de la sérotonine.
Le lithium, largement utilisé dans le traitement des troubles bipolaires, peut également, apparemment, avoir un effet positif sur les processus du métabolisme énergétique cellulaire. Il entre en compétition avec les ions sodium, participant à la régulation des pompes à calcium dans les mitochondries. A. Gardner et R. Boles, dans leur revue, citent T. Gunter, un spécialiste bien connu du métabolisme du calcium mitochondrial, qui estime que le lithium "peut affecter la vitesse à laquelle ce système s'adapte à différentes conditions et différents besoins en ATP". " De plus, le lithium est supposé réduire l'activation de la cascade apoptotique.
A. Gardner et R. Boles citent dans la revue ci-dessus de nombreuses preuves cliniques indirectes de l'effet positif des psychotropes sur les symptômes, vraisemblablement dépendants des processus de dysénergie. Ainsi, l'administration intraveineuse de chlorpromazine et d'autres antipsychotiques réduit les migraines. L'efficacité des antidépresseurs tricycliques dans le traitement de la migraine, du syndrome des vomissements cycliques et du syndrome du côlon irritable est bien connue. La carbamazépine et le valproate sont utilisés dans le traitement de la névralgie et d'autres syndromes douloureux, dont la migraine. Les inhibiteurs de la recapture du lithium et de la sérotonine sont également efficaces dans le traitement de la migraine.
En analysant les informations plutôt contradictoires données ci-dessus, nous pouvons conclure que les médicaments psychotropes sont sans aucun doute capables d'influencer les processus d'échange d'énergie cérébrale et l'activité mitochondriale. De plus, cette influence n'est pas uniquement stimulante ou inhibitrice, mais plutôt « régulatrice ». En même temps, cela peut être différent dans les neurones de différentes parties du cerveau.
Ce qui précède suggère que le manque d'énergie dans le cerveau concerne peut-être principalement des zones particulièrement touchées par le processus pathologique.
L'efficacité des médicaments énergotropes dans les troubles mentaux
Dans l'aspect du problème considéré, il est important d'obtenir la preuve d'une diminution ou d'une disparition des composantes psychopathologiques des syndromes mitochondriaux.
Dans cet aspect, le message de T. Suzuki et al., mérite l'attention en premier lieu. à propos d'un patient atteint de troubles schizophréniques sur fond de syndrome MELAS. Après l'application de coenzyme Q10 et d'acide nicotinique, le mutisme du patient a disparu pendant plusieurs jours. Il existe également un article qui rapporte le succès du dichloroacétate (souvent utilisé en "médecine mitochondriale" pour abaisser les taux de lactate) chez un homme de 19 ans atteint du syndrome MELAS, en relation avec l'effet sur l'image du délire auditif et visuel. hallucinations.
La littérature contient également une description de l'histoire d'un patient atteint du syndrome MELAS avec une mutation ponctuelle détectée 3243 mitDNA. Ce patient a développé une psychose avec des hallucinations auditives et des délires de persécution, qui a été maîtrisée en une semaine avec de faibles doses d'halopéridol. Cependant, il a ensuite développé un mutisme et une matité affective, qui n'ont pas répondu au traitement par l'halopéridol, mais ont disparu après un traitement d'un mois par l'idébénone (un analogue synthétique du coenzyme Q10) à la dose de 160 mg/jour. Chez un autre patient atteint du syndrome MELAS, la coenzyme Q10 à une dose de 70 mg/jour a aidé à faire face à la manie de persécution et au comportement agressif. Le succès de l'utilisation de la coenzyme Q10 dans le traitement du syndrome MELAS a également été indiqué dans les travaux : nous parlons d'un patient qui a non seulement empêché les épisodes de type AVC, mais également arrêté les maux de tête, les acouphènes et les épisodes psychotiques.
Il existe également des rapports sur l'efficacité de la thérapie énergétique tropique chez les patients atteints de maladie mentale. Ainsi, un patient de 23 ans souffrant de dépression résistante aux traitements a été décrit, dont la sévérité a significativement diminué après une utilisation de 2 mois de coenzyme Q10 à la dose de 90 mg par jour. Un cas similaire est décrit dans l'ouvrage. L'utilisation de carnitine en combinaison avec des cofacteurs du métabolisme énergétique s'est avérée efficace dans le traitement de l'autisme.
Ainsi, dans la littérature moderne, il existe des preuves d'un rôle important des troubles mitochondriaux dans la pathogenèse des troubles mentaux. A noter que dans cette revue, nous ne nous sommes pas attardés sur les maladies neurodégénératives du sujet âgé, pour la plupart desquelles l'importance des troubles mitochondriaux a déjà été prouvée, et leur prise en compte nécessite une publication séparée.
Sur la base des données ci-dessus, on peut affirmer que le besoin est venu de combiner les efforts des psychiatres et des spécialistes des maladies mitochondriales, visant à la fois à étudier les bases dysénergétiques des troubles de l'activité nerveuse supérieure et à analyser les manifestations psychopathologiques des maladies associées à troubles du métabolisme énergétique cellulaire. Dans cet aspect, les nouvelles approches diagnostiques (cliniques et de laboratoire) et le développement de nouvelles méthodes de traitement nécessitent une attention particulière.
1 Il convient de noter que parmi les descriptions correspondantes, une grande place est occupée par les cas avec une mutation mitDNA 3243AG détectée, une cause généralement reconnue du développement du syndrome MELAS.
Littérature