روش های الکتروشیمیایی- پویاترین ها از نظر کاربرد آنها در نظارت بر محیط زیست. متداول ترین روش های مورد استفاده در سیستم های MOS عبارتند از ولتامتری (شامل پلاروگرافی)، پتانسیومتری (شامل آیونومتری)، کولومتری و هدایت سنجی.
روش های الکتروشیمیایی تجزیه و تحلیل از وابستگی خواص الکتریکی مختلف محیط به محتوای کمی و ترکیب کیفی مواد مورد تجزیه و تحلیل استفاده می کنند:
· تغییر دادن پتانسیلالکترود بسته به فرآیندهای فیزیکی و شیمیایی رخ داده در ماده ( پتانسیومتریروش)، از جمله واکنشهای انتخابی الکترودهای انتخابی یونی که به صورت جداگانه به تعداد زیادی کاتیون و آنیون حساس هستند. یونومتریروش)؛
· تغییر دادن هدایت الکتریکی (جریان)و ثابت دی الکتریک یک ماده بسته به ماهیت محیط و غلظت اجزای آن ( هدایت سنجیو آمپرومتریکمواد و روش ها)؛
· تغییرات مقدار برقهنگامی که آنالیت وارد سلول الکتروشیمیایی می شود ( کولومتریکروش)؛
بازیابی ترکیب آنالیز شده بر روی الکترود چکاننده یا چرخان جیوه، به عنوان یک قاعده، هنگام تجزیه و تحلیل مقادیر کمی از مواد در حالت های مختلف تجمع ( پلاروگرافییا ولتامتریروش).
پلاروگراف تمام دستگاه های این گروه دارای بیشترین حساسیت برابر با 0.005-1 میکروگرم بر میلی لیتر نمونه است.
ولتامتریشامل گروهی از روش های تحلیل الکتروشیمیایی بر اساس مطالعه منحنی های پلاریزاسیون است. این روش ها هستند پلاروگرافیو تیتراسیون آمپرومتریک - دارای تنوع و تغییرات زیادی است. رایج ترین جریان پایدارپلاروگرافی.
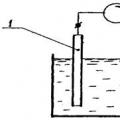
یک تاسیسات پلاروگرافی شامل یک منبع جریان مستقیم، یک تقسیم کننده ولتاژ، یک الکترود قطره ای (معمولا جیوه) یا دوار و یک الکترود کمکی (معمولاً جیوه یا سایرین) است. برای اندازه گیری جریان، یک میکرو آمپرمتر به سیستم متصل می شود. الکترودها همراه با محلول آزمایش در یک الکترولیز (سلول) قرار می گیرند.
ولتاژ اعمال شده به سلول الکترولیتی باعث پلاریزاسیون آند و کاتد می شود E= f آ- f ک +iR، جایی که من- قدرت فعلی؛ به -مقاومت محلول؛ f آو f ک- پتانسیل های آند و کاتد.
اگر مقاومت محلول را با افزودن یک الکترولیت قوی (پس زمینه) کاهش دهید، مقدار iR(افت احتمالی محلول) را می توان نادیده گرفت.
پتانسیل آند در طول عملیات سلول تقریباً ثابت می ماند، زیرا چگالی جریان کم است و سطح نسبتاً بزرگ آند قطبی نمی شود. آنگاه پتانسیل یک کاتد پلاریزه کننده چکاننده با سطح کوچک برابر خواهد بود با: E= -f ک. اغلب در اندازهگیریهای پلاروگرافی، به جای لایهای از جیوه در کف ظرف، از الکترود کالومل اشباع غیرقطبی استفاده میشود که پتانسیل آن برابر با صفر است.
داده های پلاروگرافی با اندازه گیری جریان عبوری از یک سلول الکترولیتی به عنوان تابعی از پتانسیل اعمال شده به الکترودها به دست می آید. وابستگی گرافیکی جریان به پتانسیل را موج پلاروگرافی می نامند. برنج. 2).
در ابتدای الکترولیز، در مقادیر کم EMF تحمیلی، قدرت جریان تقریباً ثابت خواهد بود و فقط به آرامی افزایش می یابد. این جریان به اصطلاح باقیمانده است که در طول الکترولیز باقی می ماند.
برنج. 2. پلاروگرام محلول 10-3 مولار کلرید روی و محلول 1 مولار کلرید پتاسیم (منحنی 1) و محلول 1 مولار کلرید پتاسیم (منحنی 2)
به محض رسیدن به پتانسیل کاهش یون (به عنوان مثال، برای یون های روی تعیین شده برابر با -1.0 V است)، تخلیه آنها با یک قطره جیوه شروع می شود:
Zn 2 + + 2 +Hg ® Zn (Hg).
یک آمالگام روی رقیق روی (Hg) در کاتد تشکیل می شود که به محض تماس قطره سقوط با آند به اجزای تشکیل دهنده آن تجزیه می شود:
روی (Hg) – 2 ® Zn 2+ +Hg.
در پتانسیل کاهش یون های روی، قدرت جریان به شدت افزایش می یابد. برنج. 2)، اما پس از رسیدن به یک مقدار مشخص، با وجود افزایش EMF اعمال شده، تقریبا ثابت می ماند. این جریان محدود کننده یا انتشار نامیده می شود، مقدار آن معمولاً متناسب با غلظت ماده تعیین شده است.
هنگام گرفتن پلاروگرام، یک الکترولیت بی تفاوت با کاتیون هایی که بسیار دشوارتر از کاتیون تجزیه و تحلیل شده کاهش می یابد، به الکترولیت مورد مطالعه اضافه می شود، به عنوان مثال، KCl، KNO 3، NH 4 Cl. در غلظت 100-1000 برابر بیشتر از غلظت ماده تعیین شده. این الکترولیت "پس زمینه" نامیده می شود. در محلول آزمایشی برای افزایش رسانایی الکتریکی و محافظت از میدان الکتریکی الکترود نشانگر (کاتد) ایجاد می شود. بنابراین، کاتیون های آنالیت توسط میدان الکتریکی کاتد جذب نمی شوند، بلکه در اثر انتشار به سمت آن حرکت می کنند.
مهمترین مشخصه پلاروگرام پتانسیل نیم موج است E 1/2 و ارتفاع موج پلاروگرافی ساعت(جریان انتشار را محدود کنید). پتانسیل نیم موج در استفاده می شود کیفیتتجزیه و تحلیل پلاروگرافی پتانسیل های نیمه موج مواد مختلف، به ترتیب افزایش ارزش منفی، به اصطلاح "طیف پلاروگرافی" را تشکیل می دهند. از آنجایی که پتانسیل نیمه موج به طور قابل توجهی به ترکیب محلول (محیط مورد تجزیه و تحلیل) بستگی دارد، پس زمینه همیشه در جداول پلاروگرافی نشان داده می شود.
که در کمیدر آنالیز پلاروگرافی از روش های نمودار کالیبراسیون، افزودنی ها، مقایسه ها و روش های محاسبه برای اندازه گیری غلظت استفاده می شود.
در میان گزینه های مختلف برای پلاروگرافی، روش پلاروگرافی پالس تفاضلی (DIP) ) برای حل مشکلات پایش محیطی، عمدتاً به دلیل حساسیت بالای آن، بیشترین تأثیر را دارد. روش DIP به شما امکان می دهد محتوای تمام مواد تعیین شده توسط پلاروگرافی کلاسیک را ارزیابی کنید. در میان سایر روش های پلاروگرافی، به ویژه برای تجزیه و تحلیل ردیابی مناسب است موج مربعپلاروگرافیکه یک حد تشخیص نزدیک به DIP را فراهم می کند، اما فقط در مورد فرآیندهای الکترود برگشت پذیر، و بنابراین از این روش اغلب برای تعیین آثار فلزات سنگین استفاده می شود. روش DIP همچنین می تواند برای تعیین سورفکتانت هایی که ظرفیت لایه الکتریکی دوگانه الکترود را تغییر می دهند، استفاده شود.
می توان از روش هایی برای تعیین ریزمحتوای یون های فلزات سنگین استفاده کرد تجزیه و تحلیل الکتروشیمیایی وارونگی (IEA) یا به روشی دیگر، تحلیل ولتامتری سلب (IVA ) که در آن فلزات تعیین شده از قبل روی الکترود رسوب می کنند و سپس در طی کنترل پلاروگرافی حل می شوند. این گزینه در ترکیب با DIP یکی از حساس ترین روش های آنالیز الکتروشیمیایی است. طراحی سختافزار IEA (IVA) نسبتاً ساده است، که امکان انجام تجزیه و تحلیلهای میدانی را فراهم میکند و ایستگاههای کنترل مداوم (مانیتورینگ) خودکار نیز میتوانند بر اساس این اصل کار کنند.
روش های IEA (IVA) تعیین یون های Cu، Pb، Bi، Sb، As، Sn In، Ga، Ag، Tl، Cd، Zn، Hg، Au، Ge، Te، Ni، Co و بسیاری از آنیون ها را فراهم می کند. مزیت مهم روش های IEA (IEA) این است (بر خلاف روش های دیگر، به عنوان مثال، مانند طیف سنجی جذب اتمی) توانایی تشخیص یون های آزاد از اشکال شیمیایی محدود آنها، که برای ارزیابی خواص فیزیکوشیمیایی مواد مورد تجزیه و تحلیل از نقطه نظر کنترل تحلیلی محیطی (مثلاً هنگام ارزیابی کیفیت آب) مهم است. بسیاری از مواد آلی را می توان با روش های IEA (IEA) پس از انباشته شدن جذب روی سطح الکترود تعیین کرد.
همچنین میتوان از روشهای پلاروگرافیک برای تعیین ذرات معلق در هوا و هوای محیطهای صنعتی پس از جذب در فیلترهای مناسب و سپس انتقال کنسانتره به محلول استفاده کرد. ترکیبات آلی موجود به شکل گازها و بخارات موجود در اتمسفر را می توان پس از جذب توسط محلول های انتخاب شده به صورت پلاروگرافیک تعیین کرد. فلزات و ترکیبات مختلف در مواد بیولوژیکی معمولاً پس از استخراج آنها به روش پلاروگرافی تعیین می شوند. تمام اندازهگیریهای پلاروگرافی، از جمله IEA (IVA)، میتوانند کاملاً خودکار باشند، که هنگام انجام آنالیزهای سریال ضروری است.
یکی از مهمترین زمینه های کاربرد پلاروگرافی، تعیین اکسیژن در آب است. برای این منظور از آشکارسازهای آمپرومتریک استفاده می شود که جریانی متناسب با غلظت اکسیژن در محلول تولید می کند.
با اعمال یک آنزیم بر روی سطح غشای آشکارساز، می توان حسگرهای آمپرومتریک آنزیمی مختلف را برای تجزیه و تحلیل های بیوشیمیایی و بالینی به دست آورد. از چنین حسگرهایی در سیستم های نظارت بر محیط زیست نیز استفاده می شود.
الکترودهایی که بر اساس اصل الکتروکاتالیستی کار می کنند برای نظارت بر گازهای مختلف (SO 2، H 2 S، CO، NO x) در هوای محل های صنعتی مناسب هستند. واکنش های الکتروشیمیایی این گازها (ایفای نقش یک کاتالیزور) که روی سطح الکترود رخ می دهد، جریانی را در سیستم الکترود ایجاد می کند که از نظر عملکردی با غلظت گازهای موجود در هوا مرتبط است.
استفاده از پلاروگرافی به تجزیه و تحلیل نمونه های گسسته محدود نمی شود و این روش به تدریج به سمت اصول آنالیز مداوم گازها و مایعات می رود.
آشکارسازهای پلاروگرافی ولتامتری با موفقیت در کروماتوگرافی مایع با کارایی بالا (HPLC) استفاده شده است. در این حالت، ترکیب یک روش جداسازی بسیار انتخابی با یک روش تشخیص حساس منجر به گسترش قابل توجهی از محدوده مواد تعیین شده توسط روش کروماتوگرافی (ردیابی مواد بسیار سمی، علف کش ها، داروها، محرک های رشد و غیره) می شود.
جزئیات روش را می توان در ادبیات تخصصی روشن کرد.
پتانسیومتری- روشی برای تعیین غلظت مواد بر اساس اندازه گیری EMF سلول های گالوانیکی برگشت پذیر.
در عمل از دو روش تحلیلی استفاده می شود: مستقیمپتانسیومتریبرای تعیین فعالیت ذرات، که می تواند با استفاده از معادله نرنست از emf سلول گالوانیکی محاسبه شود، و تیتراسیون پتانسیومتری ، که در آن تغییر در فعالیت مواد شیمیایی در طی فرآیند تیتراسیون منجر به تغییر در emf سلول گالوانیکی می شود.
تجهیزات برای انجام تیتراسیون پتانسیومتری و برای پتانسیومتری مستقیم یکسان است. مدار اندازه گیری پتانسیومتری شامل یک الکترود نشانگر و یک الکترود مرجع با پتانسیل ثابت پایدار و همچنین یک دستگاه ثانویه است. نمودار اصلی روش در نشان داده شده است برنج. 3.

1 - الکترود نشانگر؛ 2 - الکترود مرجع
برنج. 3. سلول پتانسیومتری
پتانسیل یک جفت الکترود ثابت است. تغییر غلظت آنالیت در محلول باعث تغییر EMF مدار می شود. الکترودهای نشانگر معمولاً در چهار دسته هستند انواعبسته به غشایی که محلول الکترود را از محلول آزمایشی جدا می کند: 1) الکترودهایی با غشای همگن ساخته شده از مواد پودری یا کریستالی. 2) الکترودهایی با غشای ناهمگن، که در آن ماده فعال الکترود، به عنوان مثال، در لاستیک سیلیکون توزیع می شود. 3) الکترودهایی با غشای مایع، که در آن غشاء محلولی است که روی یک ماده خنثی، به عنوان مثال، شیشه متخلخل اعمال می شود. 4) الکترودهای شیشه ای با ترکیبات شیمیایی مختلف شیشه.
الکترودهای نشانگر پتانسیل محلولی را که در آن قرار می گیرند به دست می آورند. دو تا هستند نوعالکترودهای نشانگر:
1) الکترودهای بی تفاوت (غیر قابل تخریب در طول الکترولیز).
2) الکترودهایی که در طول اندازه گیری تغییر می کنند (اکسید یا کاهش می یابند).
نقش الکترودهای بی تفاوت(اینها گاهی اوقات الکترود نامیده می شوند سومنوع) دادن یا به دست آوردن الکترون است، یعنی. رسانای الکتریسیته باشند چنین الکترودهایی می توانند از طلا، پلاتین جلا، گرافیت و مواد دیگر ساخته شوند. نمونه هایی از الکترودهای متغیر (که گاهی الکترود نامیده می شود) اولیننوع) ممکن است صفحاتی از مس، روی و سایر فلزات، و همچنین الکترودهای نشانگر کین هیدرون و هیدروژن باشد. علاوه بر این، الکترودهای نشانگر می توانند باشند الکترودهای غشایی انتخابی یونی برای تعیین کاتیون های متعدد: Li +، Pb +، Cs +، Tl +، NH +، Na +، K +، Ag + و غیره به عنوان الکترودهای مرجع ( استانداردالکترودها، که پتانسیل آن در طول اندازه گیری ثابت می ماند، رایج ترین آنها برای مثال، الکترودهای معمولی و غیر طبیعی کالومل (کالومل) با پتانسیل های 0.282+ و 0.334+ ولت و همچنین الکترود کلرید نقره اشباع شده هستند. با پتانسیل +0.201 ولت.
در یک حالت ایدهآل، اندازهگیری پتانسیومتری مستقیم EMF یک سلول گالوانیکی را میتوان از طریق معادله نرنست به فعالیت ذره تعیینشده، یا با غلظت، در صورتی که ضرایب فعالیت مربوطه مشخص باشد، مرتبط کرد:
![]()
جایی که E 0 – پتانسیل استاندارد الکترود، V; آر- ثابت گاز؛ تی- دمای مطلق؛ F –شماره فارادی؛ n- تعداد الکترون های از دست رفته یا به دست آمده؛ ، [کاهش] – غلظت های تعادلی فرم های اکسید شده و احیا شده، به ترتیب، mol/dm 3.
اگر مقادیر مرجع ثابت ها را جایگزین کرده و از لگاریتم طبیعی به اعشار حرکت کنیم، برای دمای 25 درجه سانتیگراد به دست می آید:

مهمترین شاخص در توصیف وضعیت محیط، مقدار pH این محیط است که تعیین آن ( پی اچ متری ) در حال حاضر معمولا با استفاده از الکترودهای نشانگر شیشه ای (اندازه گیری) انجام می شود. برای اندازهگیریهای طولانیمدت، طرحهای ویژهای از الکترودهای شیشهای با دستگاههای اضافی برای اطمینان از تمیز کردن غشای شیشه ایجاد شدهاند. الکترودهای شیشه ای پوشیده شده با یک غشای نیمه تراوا با یک لایه الکترولیت نیز به عنوان پایه ای برای انواع مختلف پروب ها عمل می کنند. حسگرها در تجزیه و تحلیل آب و هوا در شرایط تولید برای تعدادی از آلاینده ها (NH 3، CO 2، NO x، SO 2، H 2 S و غیره) استفاده می شود.
فرآیند در زمینه ایجاد الکترودهای انتخابی یونی (ISE) امکان نظارت بر یون های F – , I – , Br – , Cl – , CN – , SCN – , NO 3 – , NO 2 – , ClO 4 – , S 2 را می دهد. – , Na + , غلظت K + Ca 2 + ، Ag + ، Cu 2 + ، Cd 2 + ، Pb 2 + از 10-2 تا 10-7 mol/l (تقریباً 1-10-5 mg/ml) متغیر است. مانیتورینگ با استفاده از ISE با سرعت، سادگی و امکانات بیشتر برای انجام اندازهگیریهای مداوم مشخص میشود. ISEهایی توسعه یافته اند که برای طبقه وسیعی از مواد آلی، و همچنین ایزومرها در جرم، سورفکتانت ها و مواد شوینده موجود در هوای منطقه تولید و رژیم مدیریت آب شرکت های صنعتی انتخابی هستند.
پتانسیومتری همچنین در اندازه گیری پتانسیل ردوکس سیستم های مختلف ردوکس (O/R) در آب استفاده می شود. به عنوان یک قاعده، نتایج اندازه گیری با یک پتانسیل مخلوط مطابقت دارد، زیرا چندین سیستم O/W معمولاً به طور همزمان در آب وجود دارند.
لازم به ذکر است که استفاده از سنسورهای مبتنی بر اکسید فلز نیمه هادی ترانزیستورهای اثر میدانی انتخابی شیمیایی و انتخابی یونی (HSFT، ISFT) امیدوارکننده است. گزینش پذیری در این سیستم ها با انتخاب ترکیب غشاء و لایه ای که روی گیت ترانزیستور قرار گرفته است به دست می آید. سیستم در محلول مورد تجزیه و تحلیل غوطه ور می شود و اختلاف پتانسیل بین الکترود مرجع و دروازه ترانزیستور جریان جریان بین منبع و تخلیه آن را تعدیل می کند. با توجه به گزینش پذیری غشاء یا لایه رسوب شده، جریان مدوله شده تابعی از فعالیت جزء مربوط به محلول می شود. سنسورهای نیمه هادی اساس نمایشگرها و تحلیلگرهای گازها و بخارات مختلف را تشکیل می دهند. اندازه کوچک چنین سنسورهایی امکان ترکیب آنها را به شکل موزاییک روی یک بستر واحد فراهم می کند، به طوری که یک تحلیلگر به دست می آید که می تواند طیف وسیعی از مواد مضر را نظارت کند. سیگنال های حسگرهای جداگانه موجود در موزاییک را می توان به صورت متوالی و دوره ای توسط مرکز اندازه گیری سیستم تحلیلی ثبت کرد.
توسعه میکروالکترونیک امکان طراحی آنالایزرهای فشرده از نوع پروب را با استفاده از ISE های مدرن فراهم می کند. در این حالت، مداری که پاسخ جسم کنترل محیطی و حتی نمایشگر را پردازش می کند، می تواند در دسته پروب نصب شود.
در ادبیات تخصصی می توانید از جزئیات روش مطلع شوید، , , .
کولومتریکروش تجزیه و تحلیل اندازه گیری جریان واکنش الکترودی است که در آن ماده مورد مطالعه با جریان تجزیه و تحلیل شده وارد سلول کولومتری می شود. نمودار شماتیک یک سلول کولومتری در نشان داده شده است برنج. 4.

1 - محفظه کاتد؛ 2 - محفظه آند 3- میکرو آمپرمتر
برنج. 4. شماتیک یک سلول کولومتری
تجزیه و تحلیل کولومتریک بر اساس اندازه گیری مقدار الکتریسیته صرف شده برای انجام کمی فرآیند الکتروشیمیایی معین در یک نمونه معین است. مشروط بر اینکه راندمان فعلی 100% باشد. این مقدار الکتریسیته به کمک یک انتگرالگر جریان-زمان متصل به صورت سری با سلول اندازه گیری یا یک کولومتر الکترولیز است که در آن یک فرآیند الکتروشیمیایی با راندمان جریان صد درصد انجام می شود، همراه با آزاد شدن یک ماده ای که می توان مقدار آن را به راحتی و با دقت بازیابی کرد.
مطابق با قانون فارادی:
m( ایکس)/ م(ایکس) = متر(ک)/ م(ک),
جایی که متر(ایکس), m(k) -جرم ماده در حال تعیین ایکسو ماده آزاد شده در کولومتر به ترتیب. م(ایکس), م(ک) – جرم مولی معادل ماده ایکسو ماده آزاد شده در کولومتر، g/mol.

این محاسبه را می توان با استفاده از معادله توصیف قانون فارادی نیز انجام داد:
![]()
اگر قدرت جریان در طول تجزیه و تحلیل اندازه گیری شود من، الف و زمان تی، s، صرف انجام فرآیند الکتروشیمیایی می شود.
در اصلاح دیگری از این روش به نام
تیتراسیون کولومتریک
تیترانت به صورت الکترولیتی در محلول آنالیز شده در یک جریان معین تولید می شود. مصرف تیترانت در واکنش تحلیلی با باری که در محلول جریان مییابد جایگزین میشود تا زمانی که تیترنت به نقطه هم ارزی برسد.
یکی از مزایای روش های کولومتریاین است که فرآیند استانداردسازی تیترانت اغلب ضروری نیست، زیرا محاسبات بر اساس ثابت فارادی است، یعنی. این روش مطلق است و به شما امکان می دهد مقدار ماده تعیین شده و نه غلظت آن را تخمین بزنید. نقطه ضعف کولومتری با پتانسیل معین، مدت زمان فرآیند آنالیز است که با نیاز به تکمیل کامل الکترولیز همراه است. فناوری کامپیوتری با پیشبینی پایان الکترولیز با پردازش ریاضی منحنی جریان-زمان برای مراحل اولیه الکترولیز و با محاسبه مقدار الکتریسیته یا غلظت یک ماده در محلول، این زمان را کاهش میدهد. هنگام تجزیه و تحلیل نمونه های چند جزئی، می توان از آن استفاده کرد کولومتری اسکن ، که در آن پتانسیل الکترولیز به طور مداوم یا گام به گام تغییر می کند. برای چنین سیستمهایی، تیتراسیون کولومتریک به کولومتری مستقیم ترجیح داده میشود، زیرا راندمان جریان 100% در تولید تیترانت را میتوان با انتخاب صحیح معرف تیترانت و ترکیب محیط کار به راحتی به دست آورد. تیتراسیون کولومتریک برای تعیین مواد از 0.01 تا 100 میلی گرم (گاهی کمتر از 1 میکروگرم) قابل استفاده است. حجم نمونه کار معمولا از 10 تا 50 میلی لیتر است. این روش با دقت بالا مشخص می شود، خطای نسبی حتی با تیتراسیون کولومتریک محتویات میکروگرم از چند دهم درصد تجاوز نمی کند. در شرایط بهینه، تیتراسیون ها را می توان با خطای کلی بسیار کم 0.01% (مرتبط) انجام داد. اسید-بازهای مختلف، ردوکس؛ گزینه های بارش و تیتراسیون کمپلکس سنجی را می توان به صورت کولومتریک انجام داد.
آنالایزرهای گاز کولومتریک و آنالایزرهای آبی ("کولومتر") برای تعیین دی اکسید گوگرد و سولفید هیدروژن (سولفات ها و سولفیدها)، ازن (و پراکسید هیدروژن)، کلر موجود در هوا (و کلر فعال در آب) توسعه و تولید شده اند. مونوکسید کربن و دی اکسید نیتروژن در هوا (نیترات و نیتریت در آب). کولومتری همچنین به عنوان یک ابزار تشخیص الکتروشیمیایی در کروماتوگرافی مایع استفاده می شود.
جزئیات روش را می توان در ادبیات تخصصی یافت.
روش هدایت سنجیتجزیه و تحلیل بر اساس اندازه گیری هدایت الکتریکی محلول است. روش هدایت سنجی تجزیه و تحلیل شامل اندازه گیری تغییر در مقاومت محلول الکترولیت در هنگام جذب یک جزء از مخلوط است. تاسیسات هدایت سنجی، به عنوان مثال، برای تعیین مونوکسید کربن و دی اکسید، بخار بنزین، آمونیاک و غیره استفاده می شود.
رسانایی الکتریکی متقابل مقاومت است آر، ابعاد آن سانتی متر است (زیمنس) یعنی. æ = 1/ آر.
رسانایی الکتریکی یک محلول به تعداد یونها در واحد حجم محلول بستگی دارد، یعنی. روی تمرکز با، در مورد تحرک این یون ها - V.بر اساس روابط شناخته شده
![]()
جایی که ز- فاصله بین الکترودها؛ S –ناحیه الکترود؛ ک– ضریب تناسب
برای یک جفت الکترود خاص با فاصله ثابت بین آنها اس/ز= ثابت سپس
![]() ,
,
جایی که ک 1 = ک(اس/ز).
هنگام انجام محاسبات در هدایت سنجی، از مفهوم "رسانایی الکتریکی" æ 0 استفاده می شود:
![]()
در محاسبات استفاده از رسانایی الکتریکی معادل راحت است که برابر است با:
جایی که پ -تعداد مول معادل در 1 سانتی متر مکعب محلول. رسانایی الکتریکی معادل l ¥ در رقت بی نهایت برابر است با مجموع تحرکات کاتیون Uو آنیون V.
نسبت رسانایی الکتریکی معادل یک محلول الکترولیت ضعیف به رسانایی الکتریکی معادل این الکترولیت در رقت بی نهایت برابر با درجه تفکیک a این الکترولیت است:
این روش علیرغم غیر اختصاصی بودن، اغلب در سیستم های پایش محیطی در مقایسه با سایر روش های الکتروشیمیایی مورد استفاده قرار می گیرد. این با این واقعیت توضیح داده می شود که هنگام ارزیابی آلودگی، به عنوان مثال، آب و جو، نه مرحله به مرحله، بلکه کنترل خروجی (نهایی) فرآیندهای صنعتی امکان پذیر است. به دلیل رسانایی الکتریکی بسیار پایین آب، اغلب برای تخمین محتوای کل آلاینده ها کافی است، چیزی که هدایت سنجی ارائه می دهد. نمونههای معمول استفاده از روشهای هدایت سنجی در پایش محیطی، آنالیزکنندههای مواد شوینده در فاضلاب، غلظت اجزای مصنوعی در سیستمهای آبیاری و کیفیت (شوری) آب آشامیدنی است. آنالایزرهای هدایت سنجی برای پایش مداوم آلاینده های هوا و بارش مانند SO 2 و H 2 SO 4 استفاده می شود. بعلاوه هدایت سنجی مستقیممی تواند برای تعیین انواع خاصی از آلودگی استفاده شود غیر مستقیمروشهایی که تخمینهای بسیار مؤثری از محتوای مواد ذکر شده در بالا ارائه میدهند، که قبل از اندازهگیری با معرفهای خاص انتخابشده برهمکنش میکنند و تغییر ثبتشده در هدایت الکتریکی تنها به دلیل حضور محصولات مربوطه در واکنش ایجاد میشود. به این ترتیب می توانید اکسیدهای نیتروژن را پس از احیای کاتالیستی پیش آمونیاک و همچنین HCl، HBr و CO 2 را پس از واکنش اولیه با Ba(OH) 2 یا NaOH تعیین کنید. اصل توصیف شده برای تعیین CO 2 همچنین می تواند برای تعیین غیر مستقیم مواد آلی در آب استفاده شود.
علاوه بر هدایت سنجی کلاسیک، یک نسخه فرکانس بالا نیز وجود دارد ( نوسان سنجی ) که در آن سیستم الکترود نشانگر با نمونه تماس ندارد. این اصل اغلب در تحلیلگرهای هدایت پیوسته اجرا می شود.
روشهای آنالیز الکتروشیمیایی نیز در تعدادی از نشریات آموزشی و ویژه شرح داده شده است.
ادبیات
1. Drugov Yu.S., Rodin A.A.شیمی تجزیه محیطی.
سن پترزبورگ: 2002. – 464 ص.
2. پاشکویچ M.A.، Shuisky V.F. پایش محیط زیستآموزش. دانشگاه ایالتی سن پترزبورگ. – سن پترزبورگ، 2002. – 90 ص.
3. کاترال رابرت دبلیو. سنسورهای شیمیاییم.: دنیای علمی، 2000. – 144 ص.
4. Turyan Ya.I., Ruvinsky O.E., Zaitsev P.M.کاتالیزور پلاروگرافیم.: شیمی، 1998. – 272 ص.
5. بودنیکوف G.K.، Maistrenko V.N.، Murinov Yu.I. ولتامتری با الکترودهای اصلاح شده و اولترا میکرو. M.: Nauka، 1994. - 239 ص.
6. Brainina Kh.Z.، Neiman E.Ya.، Slepushkin V.V. روشهای الکتروتحلیلی وارونگیم.: 1988. – 240 ص.
7. Salikdzhanova R.F. و غیره. پلاروگراف و استفاده از آنها در تحلیل و تحقیق عملیم.: شیمی، 1988. – 192 ص.
8. Kaplan B.Ya.، Pats R.G.، Salikhdzhanova R.F. ولتامتری AC.م.: شیمی، 1985. – 264.
9. باند A.M. روش های پلاروگرافی در شیمی تجزیه. M.: شیمی، 1983.
10. افرمنکو O.A. آنالیز پتانسیومتری M.: MMA im. آنها سچنوا، 1998.
11. راهنمای مرجع برای کاربرد الکترودهای انتخابی یونی.م.: میر، 1365.
12. کوریتا I. یون ها، الکترودها، غشاها.م.: میر، 1983.
13. Nikolsky B.V.، Materova E.A. الکترودهای انتخابی یونی L.: شیمی، 1980.
14. افرمنکو O.A.تیتراسیون کولومتریک M.: MMA im. آنها سچنوا، 1990.
15. Khudyakova T.A., Koreshkov A.P. روش آنالیز هدایت سنجیکتاب درسی برای دانشگاه ها. م.: دبیرستان، 1975. – 207 ص.
16. بودنیکوف G.K.، Maistrenko V.N.، Vyaselev M.R. مبانی آنالیز الکتریکی مدرن M.: شیمی، 2000.
17. پروخورووا G.V. مقدمه ای بر روش های الکتروشیمیایی آنالیز. M.: انتشارات دانشگاه دولتی مسکو، 1991. - 97 ص.
18. روش های الکتروتحلیلی در پایش محیطی. /اد. R. Kalvoda، R. Zyka، K. Shtulik و دیگران M.: Chemistry، 1990. – 240 p.
19. پلمبک جی.روشهای تجزیه و تحلیل الکتروشیمیایی مبانی تئوری و کاربرد./ترانس. از انگلیسی م.: میر، 1365.
شرح کار
شاخه های مدرن تولید و زندگی اجتماعی مردم وظایف خاص خود را برای روش های فیزیکی و شیمیایی تجزیه و تحلیل برای کنترل کیفیت محصول ایجاد می کنند. یکی از اصلی ترین روش های فیزیکوشیمیایی آنالیز، روش های الکتروشیمیایی آنالیز است.
این روش ها می توانند به سرعت و نسبتاً دقیق بسیاری از شاخص های کیفیت محصول را تعیین کنند.
روش های الکتروشیمیایی برای تجزیه و تحلیل ترکیبات ماده به طور گسترده در صنایع مختلف استفاده می شود. آنها به شما امکان می دهند بدون توقف تولید، دریافت نتایج در مورد کیفیت محصول را خودکار کنید و تخلفات را اصلاح کنید. در صنایع غذایی، این روشها تعادل اسید و باز محصول، وجود مواد مضر و سمی و سایر شاخصها را تعیین میکنند که نه تنها بر کیفیت، بلکه بر ایمنی غذا نیز تأثیر میگذارند.
تجهیزات طراحی شده برای تجزیه و تحلیل الکتروشیمیایی نسبتا ارزان، در دسترس و آسان برای استفاده هستند. بنابراین، این روش ها نه تنها در آزمایشگاه های تخصصی، بلکه در بسیاری از صنایع به طور گسترده مورد استفاده قرار می گیرند.
در این راستا، هدف از این ku
مقدمه 2
بخش نظری 3
1.1 مشخصات کلی روشهای فیزیکوشیمیایی آنالیز 3
1.2 ویژگی های روش های الکتروشیمیایی 4
1.3 طبقه بندی روش های الکتروشیمیایی آنالیز 5
2 بخش تجربی-عملی 15
نتیجه گیری 21
مراجع 22
معرفی
فصل 1. مفاهیم کلی. طبقه بندی روش های الکتروشیمیایی تجزیه و تحلیل
فصل 2. روشهای تحلیل پتانسیومتری (پتانسیومتری)
1 اصل روش
3 تیتراسیون پتانسیومتری
فصل 3. روش هدایت سنجی تجزیه و تحلیل
1 اصل روش. مفاهیم اساسی
2 اصل هدایت سنجی
3 تیتراسیون هدایت سنجی
فصل 4. آنالیز هدایت سنجی (رسانایی سنجی)
1 ماهیت روش
2 تجزیه و تحلیل کمی پلاروگرافی
3 کاربردهای پلاروگرافی
فصل 5. تیتراسیون آمپرومتریک
فصل 6. تحلیل کولومتریک (کولومتری)
1 اصل روش
3 تیتراسیون کولومتریک
نتیجه
کتابشناسی - فهرست کتب
معرفی
روشهای الکتروشیمیایی آنالیز مجموعهای از روشهای آنالیز کمی و کیفی بر اساس پدیدههای الکتروشیمیایی هستند که در محیط مورد مطالعه یا در سطح مشترک رخ میدهند و با تغییرات در ساختار، ترکیب شیمیایی یا غلظت آنالیت مرتبط هستند.
روش های الکتروشیمیایی آنالیز به پنج گروه اصلی تقسیم می شوند: پتانسیومتری، ولتامتری، کولومتری، هدایت سنجی و آمپرومتری.
استفاده از این روش ها در تجزیه و تحلیل کمی بر اساس وابستگی مقادیر پارامترهای اندازه گیری شده در طول فرآیند الکتروشیمیایی به ماده جدا شده در محلول تجزیه شده شرکت کننده در این فرآیند الکتروشیمیایی است. چنین پارامترهایی شامل اختلاف پتانسیل الکتریکی و مقدار برق است. فرآیندهای الکتروشیمیایی فرآیندهایی هستند که به طور همزمان با یک واکنش شیمیایی و تغییر در خواص الکتریکی سیستم همراه هستند که در چنین مواردی می توان آن را یک سیستم الکتروشیمیایی نامید. در عمل تحلیلی، یک سیستم الکتروشیمیایی معمولاً حاوی یک سلول الکتروشیمیایی است که شامل یک ظرف حاوی محلول آزمایشی رسانای الکتریکی است که الکترودها در آن غوطه ور می شوند.
روش های الکتروشیمیایی مستقیم و غیر مستقیم وجود دارد. در روش های مستقیم از وابستگی قدرت جریان (پتانسیل و ...) به غلظت جزء در حال تعیین استفاده می شود. در روشهای غیرمستقیم، قدرت جریان (پتانسیل و ...) اندازهگیری میشود تا نقطه پایانی تیتراسیون جزء در حال تعیین با تیتر مناسب پیدا شود، یعنی از وابستگی پارامتر اندازهگیری شده به حجم تیتر استفاده شود.
فصل 1. مفاهیم کلی. طبقه بندی روش های آنالیز الکتروشیمیایی
شیمی الکتروتحلیلی شامل روش های الکتروشیمیایی تجزیه و تحلیل بر اساس واکنش های الکترودی و انتقال الکتریسیته از طریق محلول ها است.
استفاده از روش های الکتروشیمیایی در تجزیه و تحلیل کمی مبتنی بر استفاده از وابستگی مقادیر پارامترهای اندازه گیری شده فرآیندهای الکتروشیمیایی (تفاوت پتانسیل الکتریکی، جریان، مقدار الکتریسیته) بر محتوای آنالیت در محلول تجزیه و تحلیل شده است. این فرآیند الکتروشیمیایی فرآیندهای الکتروشیمیایی به فرآیندهایی گفته می شود که با وقوع همزمان واکنش های شیمیایی و تغییر خواص الکتریکی سیستم همراه است که در چنین مواردی می توان آن را سیستم الکتروشیمیایی نامید. در عمل تحلیلی، یک سیستم الکتروشیمیایی معمولاً حاوی یک سلول الکتروشیمیایی، از جمله ظرفی با محلول آزمایشی رسانای الکتریکی است که الکترودها در آن غوطه ور می شوند.
طبقه بندی روش های الکتروشیمیایی تجزیه و تحلیل. روش های الکتروشیمیایی تجزیه و تحلیل به روش های مختلفی طبقه بندی می شوند طبقه بندی بر اساس در نظر گرفتن ماهیت منبع انرژی الکتریکی در سیستم است. دو گروه روش وجود دارد:
الف) روشهای بدون تحمیل پتانسیل خارجی (خارجی).
منبع انرژی الکتریکی خود سیستم الکتروشیمیایی است که یک عنصر گالوانیکی (مدار گالوانیکی) است. این روش ها شامل روش های پتانسیومتری می باشد. نیروی الکتروموتور - EMF - و پتانسیل الکترود در چنین سیستمی به محتوای آنالیت در محلول بستگی دارد.
ب) روش هایی با تحمیل پتانسیل خارجی (خارجی). این روش ها عبارتند از:
تجزیه و تحلیل هدایت سنجی - بر اساس اندازه گیری هدایت الکتریکی محلول ها به عنوان تابعی از غلظت آنها.
تجزیه و تحلیل ولتامتری - بر اساس اندازه گیری جریان به عنوان تابعی از تفاوت پتانسیل شناخته شده اعمال شده و غلظت محلول.
تجزیه و تحلیل کولومتریک - بر اساس اندازه گیری مقدار برق عبوری از محلول به عنوان تابعی از غلظت آن.
تجزیه و تحلیل الکتروگراویمتری - بر اساس اندازه گیری جرم محصول یک واکنش الکتروشیمیایی.
طبقه بندی بر اساس روش استفاده از روش های الکتروشیمیایی. روش های مستقیم و غیر مستقیم وجود دارد.
الف) روشهای مستقیم پارامتر الکتروشیمیایی به عنوان یک تابع شناخته شده از غلظت محلول اندازه گیری می شود و با توجه به قرائت دستگاه اندازه گیری مربوطه، محتوای ماده تعیین شده در محلول پیدا می شود.
ب) روشهای غیرمستقیم روشهای تیتراسیون هستند که در آنها پایان تیتراسیون بر اساس اندازه گیری پارامترهای الکتریکی سیستم تعیین می شود.
مطابق با این طبقه بندی، برای مثال، بین هدایت سنجی مستقیم و تیتراسیون هدایت سنجی تفاوت قائل می شود.
فصل 2. روش آنالیز پتانسیومتری (پتانسیومتری)
1 اصل روش
آنالیز پتانسیومتری (پتانسیومتری) بر اساس اندازه گیری پتانسیل emf و الکترود به عنوان تابعی از غلظت محلول آنالیز شده است.
اگر در یک سیستم الکتروشیمیایی - در یک سلول گالوانیکی - واکنشی روی الکترودها رخ دهد:
aA+bB↔dD + eE
با انتقال n الکترون، معادله نرنست برای emf E این واکنش به شکل زیر است:
E꞊E˚- RTnFlnaDda Eea(A)a aBb
که طبق معمول E° EMF استاندارد واکنش است (تفاوت در پتانسیل های الکترود استاندارد)، R ثابت گاز، T دمای مطلقی است که واکنش در آن رخ می دهد، F عدد فارادی است. a (A)، a (B)، a (D) و i (E) - فعالیت های معرف های شرکت کننده در واکنش. معادله (10.1) برای emf یک سلول گالوانیکی با عملکرد برگشت پذیر معتبر است.
برای دمای اتاق، معادله (10.1) را می توان به شکل زیر نشان داد:
E꞊E˚- 0.059nlnaDda Eea(A)a aBb
در شرایطی که فعالیت معرف ها تقریباً برابر با غلظت آنها باشد، رابطه (1) به معادله (3) تبدیل می شود:
꞊E˚- RTnFlncDdc EecAa aBb
که در آن c(A)، c(B)، c(E)، c(D) غلظت معرف ها هستند. برای دمای اتاق، این معادله را می توان به صورت (4) نشان داد:
꞊E˚- 0.059nlncDdc EecAa aBb
برای اندازه گیری پتانسیومتری، دو الکترود در یک سلول الکتروشیمیایی استفاده می شود: یک الکترود نشانگر، که پتانسیل آن به غلظت ماده آنالیت (تعیین کننده پتانسیل) در محلول آنالیز شده بستگی دارد، و یک الکترود مرجع، که پتانسیل آن ثابت می ماند. تحت شرایط تجزیه و تحلیل بنابراین، مقدار EMF که با معادلات (1)-(4) تعیین می شود، می تواند به عنوان اختلاف بین پتانسیل واقعی این دو الکترود محاسبه شود.
در پتانسیومتری از انواع الکترودهای زیر استفاده می شود: الکترودهای نوع اول، دوم، ردوکس، الکترودهای غشایی.
الکترودهای نوع اول الکترودهایی هستند که توسط یک کاتیون مشترک با مواد الکترود برگشت پذیر هستند. سه نوع الکترود از نوع اول وجود دارد.
الف) فلز M غوطه ور در محلول نمکی از همان فلز. یک واکنش برگشت پذیر در سطح چنین الکترودهایی رخ می دهد:
Mn+ + ne = M
پتانسیل واقعی چنین الکترودی از نوع اول به فعالیت a(Mn+) کاتیون های فلزی بستگی دارد و با معادلات (5)-(8) توصیف می شود.
به طور کلی، برای هر درجه حرارت:
꞊E˚+ RTnFln a (Mn+)
برای دمای اتاق:
꞊E˚+ 0.059nln a (Mn+)
در غلظت های پایین c(Mn+)، زمانی که فعالیت کاتیون های فلزی a(Mn+) تقریباً برابر با غلظت آنها باشد:
꞊E˚+ RTnFln c(Mn+)
برای دمای اتاق:
ب) الکترودهای گاز، به عنوان مثال، الکترود هیدروژن، از جمله الکترود هیدروژن استاندارد. پتانسیل یک الکترود هیدروژن گازی با عملکرد برگشت پذیر با فعالیت یون های هیدروژن تعیین می شود. مقدار pH محلول و در دمای اتاق برابر است با:
꞊E˚+ 0.059 lg a (H30+) = 0.059 lg a (H3O+) = -0.059рН
از آنجایی که برای الکترود هیدروژن پتانسیل استاندارد صفر در نظر گرفته می شود ( £° =0), و مطابق با واکنش الکترود: H++e = N تعداد الکترون های شرکت کننده در این واکنش برابر با یک است: n = 1. ج) الکترودهای آمالگام که یک آمالگام فلزی غوطه ور در محلولی حاوی کاتیون های همان فلز هستند. پتانسیل چنین الکترودهایی از نوع اول به فعالیت کاتیون های فلزی a(Mn+) در محلول و فعالیت فلز a(M) در آمالگام بستگی دارد: ꞊E˚+ RTnFlna(Mn+)a(M) الکترودهای آمالگام بسیار برگشت پذیر هستند. الکترودهای نوع دوم آنیون برگشت پذیر هستند. انواع زیر از الکترودهای نوع دوم متمایز می شوند. الف) فلزی که سطح آن با نمک کم محلول همان فلز، غوطه ور در محلولی حاوی آنیون های تشکیل دهنده این نمک کم محلول پوشیده شده است. به عنوان مثال، الکترود کلرید نقره Ag|AgCl، KS1 یا الکترود کالومل Hg|Hg2Cl2، KS1 است. یک الکترود کلرید نقره شامل یک سیم نقره ای است که با نمک کمی محلول در آب، AgCI، پوشیده شده و در محلول آبی کلرید پتاسیم غوطه ور شده است. یک واکنش برگشت پذیر در الکترود کلرید نقره رخ می دهد الکترود کالومل شامل جیوه فلزی است که با خمیری از جیوه کم محلول (1) کلرید Hg2Cl2 - کالومل، در تماس با محلول آبی کلرید پتاسیم پوشیده شده است. یک واکنش برگشت پذیر در الکترود کالومل رخ می دهد: Cl2 + 2e = 2Hg + 2SG. پتانسیل واقعی الکترودهای نوع دوم به فعالیت آنیون ها و الکترود برگشت پذیری که واکنش روی آن انجام می شود بستگی دارد: Ne = M + An- توصیف شده توسط معادلات نرنست (9)-(12). به طور کلی، در هر دمای قابل قبول T: ꞊E˚- RTnFln a(An-) برای دمای اتاق: ꞊E˚- 0.059nln a(An-) برای شرایطی که فعالیت آنیون ها تقریباً برابر با غلظت آنها c(A"~ است): E꞊E˚- RTnFln c(An-) برای دمای اتاق: ꞊E˚- 0.059nln c(An-) به عنوان مثال، پتانسیل واقعی E1 و E2 الکترودهای کلرید نقره و کلومل به ترتیب در دمای اتاق را می توان به صورت زیر نشان داد: ꞊E1˚- 0.0591g a (Cl-)،꞊E2˚- 0.0591g a (Cl-). الکترودهای نوع دوم در عملکرد بسیار برگشت پذیر و پایدار هستند، بنابراین اغلب به عنوان الکترودهای مرجع استفاده می شوند که قادر به حفظ پایدار مقدار پتانسیل ثابت هستند. ب) الکترودهای گاز نوع دوم، مثلاً الکترود کلر Pt، Cl2 KS1. الکترودهای گاز نوع دوم به ندرت در تجزیه و تحلیل پتانسیومتری کمی استفاده می شوند. الکترودهای ردوکس از یک ماده خنثی (پلاتین، طلا، تنگستن، تیتانیوم، گرافیت و غیره) تشکیل شدهاند که در محلولی حاوی Ox اکسید شده و اشکال قرمز کاهش یافته این ماده غوطهور شدهاند. دو نوع الکترود ردوکس وجود دارد: الف) الکترودهایی که پتانسیل آنها به فعالیت یونهای هیدروژن بستگی ندارد، به عنوان مثال، Pt | FeCl3، FeCI2، Pt | K3، K4، و غیره؛ ب) الکترودهایی که پتانسیل آنها به فعالیت یونهای هیدروژن بستگی دارد، مثلاً الکترود کین هیدرون. در الکترود ردوکس، که پتانسیل آن به فعالیت یون های هیدروژن بستگی ندارد، یک واکنش برگشت پذیر رخ می دهد: گاو + ن = قرمز پتانسیل واقعی چنین الکترود ردوکس به فعالیت اشکال اکسید شده و احیا شده یک ماده داده شده بستگی دارد و برای یک الکترود با عملکرد برگشت پذیر، بسته به شرایط (بر اساس قیاس با پتانسیل های مورد بحث در بالا)، با معادلات نرنست توصیف می شود. 13)-(16): ꞊E˚+ RTnFln a (Ox)a (قرمز)꞊E˚+ 0.059nlg a (Ox)a (قرمز)꞊E˚+ RTnFln c(Ox)c (قرمز)꞊E˚+ 0.059nlg c (Ox) ج (قرمز) اگر یون های هیدروژن در واکنش الکترود شرکت کنند، فعالیت (غلظت) آنها در معادلات نرنست مربوطه برای هر مورد خاص در نظر گرفته می شود. الکترودهای غشایی یا یون انتخابی الکترودهایی هستند که برای یونهای خاصی (کاتیونها یا آنیونها) که توسط غشای جامد یا مایع جذب میشوند، برگشتپذیر هستند. پتانسیل واقعی چنین الکترودهایی به فعالیت آن یونهای موجود در محلول بستگی دارد که توسط غشاء جذب می شوند. الکترودهای غشایی جامد حاوی یک غشای بسیار نازک هستند که در دو طرف آن محلولهای مختلفی حاوی یونهای مشابه، اما با غلظتهای متفاوت وجود دارد: یک محلول (استاندارد) با غلظت دقیق مشخص شده از یونهایی که باید تعیین شوند. محلولی که باید با غلظت نامعلومی از یونهای تعیینشده آنالیز شود. به دلیل غلظتهای متفاوت یونها در هر دو محلول، یونهای طرفهای مختلف غشاء به مقدار نابرابر جذب میشوند و بار الکتریکی ناشی از جذب یونها در طرفهای مختلف غشا نیز متفاوت است. در نتیجه، اختلاف پتانسیل غشایی ایجاد می شود. تعیین یون ها با استفاده از الکترودهای انتخاب کننده یون غشایی را یونومتری می گویند. همانطور که در بالا ذکر شد، در اندازه گیری های پتانسیومتری، سلول الکتروشیمیایی شامل دو الکترود - یک الکترود نشانگر و یک الکترود مرجع است. بزرگی EMF تولید شده در سلول برابر است با اختلاف پتانسیل بین این دو الکترود. از آنجایی که پتانسیل الکترود مرجع تحت شرایط تعیین پتانسیومتری ثابت می ماند، emf فقط به پتانسیل الکترود نشانگر بستگی دارد، یعنی. در مورد فعالیت (غلظت) یونهای خاص در محلول. این مبنایی برای تعیین پتانسیومتری غلظت یک ماده معین در محلول آنالیز شده است. برای تعیین پتانسیومتری غلظت یک ماده در محلول، هم از پتانسیومتری مستقیم و هم تیتراسیون پتانسیومتری استفاده می شود، اگرچه روش دوم بسیار بیشتر از روش اول استفاده می شود. تعیین غلظت یک ماده در پتانسیومتری مستقیم معمولاً با استفاده از روش منحنی کالیبراسیون یا روش افزودن استاندارد انجام می شود. الف) روش گراف کالیبراسیون. یک سری 5-7 محلول استاندارد با محتوای مشخص آنالیت تهیه کنید. غلظت آنالیت و قدرت یونی در محلول های استاندارد نباید تفاوت زیادی با غلظت و قدرت یونی محلول آنالیز شده داشته باشد: در این شرایط، خطاهای تعیین کاهش می یابد. قدرت یونی همه محلول ها با وارد کردن یک الکترولیت بی تفاوت ثابت نگه داشته می شود. محلول های استاندارد به طور متوالی به یک سلول الکتروشیمیایی (پتانسیومتری) وارد می شوند. به طور معمول این سلول یک لیوان شیشه ای است که در آن یک الکترود نشانگر و یک الکترود مرجع قرار می گیرد. EMF محلول های استاندارد با شستشوی کامل الکترودها و شیشه با آب مقطر قبل از پر کردن سلول با هر محلول استاندارد اندازه گیری می شود. بر اساس داده های به دست آمده، یک نمودار کالیبراسیون در مختصات EMF-log c ساخته می شود، که در آن c غلظت آنالیت در محلول استاندارد است. به طور معمول این نمودار یک خط مستقیم است. سپس محلول آنالیز شده به سلول الکتروشیمیایی اضافه می شود (پس از شستشوی سلول با آب مقطر) و emf سلول اندازه گیری می شود. با استفاده از نمودار کالیبراسیون، log c(X) یافت می شود، که در آن c(X) غلظت آنالیت در محلول آنالیز شده است. ب) روش جمع استاندارد. حجم مشخصی V(X) از محلول آنالیز شده با غلظت c(X) به سلول الکتروشیمیایی اضافه می شود و emf سلول اندازه گیری می شود. سپس، یک حجم کوچک با دقت اندازه گیری شده از محلول استاندارد V(st) با غلظت شناخته شده و به اندازه کافی بزرگ c(st) آنالیت به همان محلول اضافه می شود و emf سلول دوباره تعیین می شود. غلظت c(X) آنالیت را در محلول آنالیز شده با استفاده از فرمول (10.17) محاسبه کنید: c(X)= c(st) V (st)V X+ V (st) جایی که △ E تفاوت بین دو مقدار EMF اندازه گیری شده است، n تعداد الکترون های شرکت کننده در واکنش الکترود است. استفاده از پتانسیومتری مستقیم این روش برای تعیین غلظت یون های هیدروژن (pH محلول ها)، آنیون ها و یون های فلزی (یونومتری) استفاده می شود. هنگام استفاده از پتانسیومتری مستقیم، انتخاب یک الکترود نشانگر مناسب و اندازه گیری دقیق پتانسیل تعادل نقش مهمی ایفا می کند. هنگام تعیین pH محلول ها، از الکترودها به عنوان الکترودهای شاخص استفاده می شود که پتانسیل آن به غلظت یون های هیدروژن بستگی دارد: شیشه، هیدروژن، کین هیدرون و برخی دیگر. الکترود شیشه ای غشایی که در یون های هیدروژن برگشت پذیر است بیشتر مورد استفاده قرار می گیرد. پتانسیل چنین الکترود شیشه ای با غلظت یون های هیدروژن تعیین می شود، بنابراین EMF یک مدار شامل یک الکترود شیشه ای به عنوان یک نشانگر در دمای اتاق با این معادله توصیف می شود: K + 0.059rN، که در آن K ثابت به مواد غشا و ماهیت الکترود مرجع بستگی دارد. الکترود شیشه ای به شما امکان می دهد PH را در محدوده pH = 0-10 (اغلب در محدوده pH = 2-10) تعیین کنید و در عملکرد بسیار برگشت پذیر و پایدار است. الکترود کین هیدرون که اغلب در گذشته استفاده می شد، یک الکترود ردوکس است که پتانسیل آن به غلظت یون های هیدروژن بستگی دارد. این شامل یک سیم پلاتین غوطه ور در محلول اسید (معمولا HC1) اشباع شده با کین هیدرون، یک ترکیب هم مولکولی از کینون و هیدروکینون با ترکیب C6H402 است. C6H4(OH)2 (پودر سبز تیره، کمی محلول در آب). تعیین شماتیک الکترود کین هیدرون: Pt | کوین هیدرون، HC1. یک واکنش ردوکس در الکترود کین هیدرون رخ می دهد: C6H402 + 2H+ + 2e = C6H4(OH)2 پتانسیل الکترود کین هیدرون در دمای اتاق با فرمول توصیف می شود E°-0.059рН. الکترود کوین هیدرون به شما امکان می دهد PH محلول ها را در محدوده pH = 0-8.5 اندازه گیری کنید. در pH< 0 хингидрон гидролитически расщепляется: при рН >8.5 هیدروکینون که یک اسید ضعیف است تحت واکنش خنثی سازی قرار می گیرد الکترود کین هیدرون را نمی توان در حضور عوامل اکسید کننده و کاهنده قوی استفاده کرد. همانطور که در بالا ذکر شد، الکترودهای انتخابی یون غشایی در یونومتری به عنوان شاخص برای تعیین کاتیون های مختلف (Li+، Na+، K+ Mg2t، Ca2+، Cd2+، Fe2+، Ni2+ و غیره) یون ها (F-، Cl-، Br-، I-، S2-، و غیره). از مزایای پتانسیومتری مستقیم می توان به سادگی و سرعت اندازه گیری ها اشاره کرد؛ اندازه گیری ها به حجم کمی از محلول ها نیاز دارند. 3 تیتراسیون پتانیومتری تیتراسیون پتانسیومتری روشی برای تعیین حجم تیترانت صرف شده برای تیتراسیون آنالیت در محلول تجزیه و تحلیل شده با اندازه گیری EMF (در طول فرآیند تیتراسیون) با استفاده از یک مدار گالوانیکی متشکل از یک الکترود نشانگر است. و یک الکترود مرجع در تیتراسیون پتانسیومتری، محلول مورد تجزیه و تحلیل واقع در یک سلول الکتروشیمیایی تیتر می شود یک تیترانت مناسب، پایان تیتراسیون را با تغییر شدید در EMF مدار اندازه گیری شده ثابت می کند - پتانسیل الکترود نشانگر، که به غلظت یون های مربوطه بستگی دارد و در نقطه هم ارزی به شدت تغییر می کند. تغییر در پتانسیل الکترود نشانگر در طول فرآیند تیتراسیون بسته به حجم تیتر اضافه شده اندازه گیری می شود. بر اساس دادههای بهدستآمده، منحنی تیتراسیون پتانسیومتری ساخته شده و حجم تیترانت مصرفی در پیل سوختی از این منحنی تعیین میشود. تیتراسیون پتانسیومتری نیازی به استفاده از نشانگرهایی ندارد که در نزدیکی پیل سوختی تغییر رنگ می دهند. کاربرد تیتراسیون پتانسیومتری این روش جهانی است؛ می توان از آن برای نشان دادن پایان تیتراسیون در همه انواع تیتراسیون استفاده کرد: اسید-باز، ردوکس، کمپلکس سنجی، بارش، و هنگام تیتراسیون در محیط های غیر آبی. الکترودهای شیشه، جیوه، انتخاب کننده یون، پلاتین و نقره به عنوان الکترودهای نشانگر و الکترودهای کالومل، کلرید نقره و شیشه به عنوان الکترودهای مرجع استفاده می شوند. این روش دقت و حساسیت بالایی دارد: تیتراسیون را در محیطهای کدر، رنگی، غیرآبی و تعیین جداگانه اجزای مخلوط در یک محلول آنالیز شده، برای مثال، تعیین جداگانه یونهای کلرید و یدید در طی تیتراسیون آرژانتومتری امکانپذیر میکند. روش های تیتراسیون پتانسیومتری برای تجزیه و تحلیل بسیاری از مواد دارویی مانند اسید اسکوربیک، داروهای سولفا، باربیتورات ها، آلکالوئیدها و غیره استفاده می شود. بنیانگذار آنالیز هدایت سنجی را فیزیکدان و شیمیدان فیزیک آلمانی F.V.G. Kohlrausch (1840-1910)، که برای اولین بار در سال 1885 معادله ای را پیشنهاد کرد که رابطه ای بین رسانایی الکتریکی محلول های الکترولیت های قوی و غلظت آنها ایجاد می کند. که در اواسط دهه 40 قرن XX یک روش تیتراسیون هدایت سنجی فرکانس بالا توسعه داده شد. از ابتدای دهه 60. قرن XX آشکارسازهای هدایت سنجی در کروماتوگرافی مایع شروع به استفاده کردند. 1 اصل روش. مفاهیم اساسی آنالیز هدایت سنجی (رسانایی سنجی) بر اساس استفاده از رابطه بین هدایت الکتریکی (رسانایی الکتریکی) محلول های الکترولیت و غلظت آنها است. رسانایی الکتریکی محلول های الکترولیت - هادی های نوع دوم - بر اساس اندازه گیری مقاومت الکتریکی آنها در یک سلول الکتروشیمیایی که یک ظرف شیشه ای (شیشه ای) با دو الکترود لحیم شده در آن است که محلول الکترولیت آزمایشی بین آنها قرار می گیرد، قضاوت می شود. واقع شده. یک جریان الکتریکی متناوب از سلول عبور می کند. الکترودها اغلب از فلز پلاتین ساخته می شوند که برای افزایش سطح الکترودها، با رسوب الکتروشیمیایی ترکیبات پلاتین از محلول ها (الکترودهای پلاتین شده پلاتین) با لایه ای از پلاتین اسفنجی پوشانده می شود. برای جلوگیری از عوارض مرتبط با فرآیندهای الکترولیز و پلاریزاسیون، اندازه گیری های هدایت سنجی در یک میدان الکتریکی متناوب انجام می شود. مقاومت الکتریکی R لایه محلول الکترولیت بین الکترودها، مانند مقاومت الکتریکی رساناهای نوع اول، با طول (ضخامت) l این لایه نسبت مستقیم و با سطح S الکترودها نسبت معکوس دارد: R= ρ lS lkS که در آن ضریب تناسب p مقاومت الکتریکی خاص و مقدار معکوس k = 1/p رسانایی الکتریکی خاص (رسانایی الکتریکی) نامیده می شود. از آنجایی که مقاومت الکتریکی R بر حسب اهم اندازه گیری می شود، ضخامت l لایه محلول الکترولیت بر حسب سانتی متر و سطح S الکترودها بر حسب سانتی متر مربع است، هدایت الکتریکی ویژه k بر حسب واحد اهم-1 اندازه گیری می شود. cm-1، یا، از آنجایی که اهم-1 زیمنس (Sm) است، سپس - در واحدهای Sm سانتی متر-1. در معنای فیزیکی، رسانایی الکتریکی ویژه، رسانایی الکتریکی یک لایه الکترولیت است که بین دو طرف یک مکعب به طول ضلع 1 سانتیمتر، از نظر عددی برابر با جریان عبوری از لایهای از محلول الکترولیت با سطح مقطع است. 1 سانتی متر مربع با گرادیان پتانسیل الکتریکی اعمال شده 1 V/cm. هدایت الکتریکی خاص به ماهیت الکترولیت و حلال، غلظت محلول و دما بستگی دارد. با افزایش غلظت محلول الکترولیت، هدایت الکتریکی ویژه آن ابتدا افزایش می یابد، سپس از حداکثر عبور می کند و سپس کاهش می یابد. این ماهیت تغییر در هدایت الکتریکی به دلایل زیر است. در ابتدا، با افزایش غلظت الکترولیت، تعداد یون ها - ذرات حامل جریان - برای الکترولیت های قوی و ضعیف افزایش می یابد. بنابراین رسانایی الکتریکی محلول (جریان الکتریکی عبوری از آن) افزایش می یابد. سپس با افزایش غلظت محلول، ویسکوزیته آن (کاهش سرعت حرکت یون ها) و برهمکنش های الکترواستاتیکی بین یون ها افزایش می یابد که از افزایش جریان الکتریکی جلوگیری می کند و در غلظت های کافی به کاهش آن کمک می کند. در محلول های الکترولیت های ضعیف، با افزایش غلظت، درجه تفکیک مولکول های الکترولیت کاهش می یابد که منجر به کاهش تعداد یون ها - ذرات رسانا - و کاهش رسانایی الکتریکی ویژه می شود. در محلولهای الکترولیتهای قوی در غلظتهای بالا، تشکیل پیوندهای یونی (دوقلوهای یونی، سه راهیها و غیره) امکانپذیر است که به کاهش رسانایی الکتریکی نیز کمک میکند. هدایت الکتریکی ویژه محلول های الکترولیت با افزایش دما به دلیل کاهش ویسکوزیته محلول ها افزایش می یابد که منجر به افزایش سرعت حرکت یون ها می شود و برای الکترولیت های ضعیف نیز باعث افزایش درجه یونیزاسیون آنها می شود. (تجزیه به یون). بنابراین، اندازهگیریهای کمی هدایت سنجی باید در دمای ثابت انجام شوند و سلول هدایت سنجی را ترموستات کنند. علاوه بر رسانایی الکتریکی خاص، هدایت سنجی از رسانایی الکتریکی معادل X و رسانایی الکتریکی مولی p استفاده می کند. از نظر فیزیکی، رسانایی الکتریکی معادل X، رسانایی الکتریکی لایه ای از محلول الکترولیت به ضخامت 1 سانتی متر است که بین الکترودهای یکسان قرار دارد و دارای مساحتی است که حجم محلول الکترولیت محصور در بین آنها حاوی 1 گرم equiv از ماده محلول است. در این مورد، جرم مولی معادل، جرم مولی ذرات یکسان با یک عدد بار واحد ("بار") در نظر گرفته می شود، به عنوان مثال، H+، Br -، 12Ca2+، 13Fe3+ و غیره. رسانایی الکتریکی معادل با کاهش غلظت محلول الکترولیت افزایش می یابد. حداکثر مقدار رسانایی الکتریکی معادل با رقت بی نهایت محلول به دست می آید. هدایت الکتریکی معادل، مانند رسانایی ویژه، با افزایش دما افزایش می یابد. رسانایی الکتریکی معادل X با رسانایی الکتریکی خاص k با رابطه (20) مرتبط است: λ= 1000 kc در هدایت سنجی مستقیم، غلظت یک ماده در محلول مورد تجزیه و تحلیل از نتایج اندازه گیری رسانایی الکتریکی ویژه این محلول تعیین می شود. هنگام پردازش داده های اندازه گیری، از دو روش استفاده می شود: روش محاسبه و روش نمودار کالیبراسیون. روش محاسبه. مطابق با رابطه (10.20)، اگر رسانایی الکتریکی خاص k و رسانایی الکتریکی معادل شناخته شده باشند، می توان غلظت مولی معادل c الکترولیت در محلول را محاسبه کرد. : c = 1000 kλ هدایت الکتریکی خاص به صورت تجربی بر اساس اندازه گیری مقاومت الکتریکی یک سلول هدایت سنجی ترموستات تعیین می شود. رسانایی الکتریکی معادل محلول λ برابر با مجموع تحرکات کاتیونی است λ+ و آنیون X λ -:
λ = λ + + λ-
اگر تحرک کاتیون و آنیون مشخص باشد، می توان غلظت را با استفاده از فرمول (24) محاسبه کرد: c = 1000 kλ + + λ- این کار هنگام تعیین غلظت الکترولیت کم محلول در محلول اشباع آن (کلسیم، سولفات باریم، هالیدهای نقره و غیره) توسط هدایت سنجی مستقیم انجام می شود. روش گراف کالیبراسیون مجموعهای از محلولهای استاندارد تهیه میشود که هر کدام حاوی غلظت مشخصی از آنالیت هستند و هدایت الکتریکی آنها در دمای ثابت در یک سلول هدایت سنجی ترموستات اندازهگیری میشود. بر اساس دادههای بهدستآمده، یک نمودار کالیبراسیون ساخته میشود که غلظت محلولهای استاندارد را روی محور آبسیسا و مقادیر رسانایی الکتریکی خاص در امتداد محور ارتین ترسیم میکند. مطابق با رابطه (24)، نمودار رسم شده معمولاً یک خط مستقیم را در محدوده نسبتاً کمی از تغییرات غلظت نشان می دهد. در طیف وسیعی از غلظتها، زمانی که تحرک کاتیون و آنیون موجود در رابطه (24) میتواند بهطور محسوسی تغییر کند، انحراف از وابستگی خطی مشاهده میشود. سپس، دقیقاً تحت شرایط یکسان، هدایت الکتریکی ویژه k(X) الکترولیت تعیین شده در محلول آنالیز شده با غلظت نامعلوم c(X) اندازه گیری می شود و مقدار مورد نظر c(X) از نمودار بدست می آید. به عنوان مثال، محتوای باریم در آب باریت - محلول اشباع شده هیدروکسید باریم - به این ترتیب تعیین می شود. کاربرد هدایت سنجی مستقیم روش هدایت سنجی مستقیم با سادگی و حساسیت بالا مشخص می شود. با این حال، روش بسیار انتخابی نیست. هدایت سنجی مستقیم کاربرد محدودی در تحلیل دارد. برای تعیین حلالیت الکترولیت های کم محلول، کنترل کیفیت آب مقطر و محصولات غذایی مایع (شیر، نوشیدنی ها و غیره)، تعیین میزان نمک کل در آب معدنی، دریا، رودخانه و در برخی موارد دیگر استفاده می شود. . 3 تیتراسیون هدایت سنجی در تیتراسیون هدایت سنجی، پیشرفت تیتراسیون با تغییر در رسانایی الکتریکی محلول مورد تجزیه و تحلیل واقع در یک سلول هدایت سنجی بین دو الکترود بی اثر (معمولاً از پلاتین پلاتین شده) نظارت می شود. بر اساس دادههای بهدستآمده، یک منحنی تیتراسیون هدایت سنجی ترسیم میشود که وابستگی هدایت الکتریکی محلول تیتر شده را به حجم تیتر اضافه شده منعکس میکند. نقطه پایان تیتراسیون اغلب با برون یابی بخش هایی از منحنی تیتراسیون در ناحیه ای که شیب آن تغییر می کند پیدا می شود.در این حالت استفاده از نشانگرهایی که در نزدیکی TE تغییر رنگ می دهند لازم نیست. در تیتراسیون هدایت سنجی، انواع مختلفی از واکنش ها استفاده می شود: فرآیندهای اسید-باز، ردوکس، رسوب، فرآیندهای کمپلکس. کاربرد تیتراسیون هدایت سنجی. روش تیتراسیون هدایت سنجی دارای چندین مزیت است. تیتراسیون را می توان در محیط های کدر، رنگی و مات انجام داد. حساسیت روش بسیار بالا است - تا ~10~* mol/l. محدوده خطای تعیین از 0.1 تا 2٪ است. تجزیه و تحلیل می تواند خودکار باشد. از معایب روش می توان به گزینش پذیری کم اشاره کرد. مفهوم تیتراسیون هدایت سنجی فرکانس بالا (فرکانس رادیویی). پیشرفت تیتراسیون با استفاده از تکنیک هدایت سنجی جریان متناوب اصلاح شده، که در آن فرکانس جریان متناوب می تواند به مرتبه یک میلیون نوسان در ثانیه برسد، نظارت می شود. به طور معمول، الکترودها در قسمت بیرونی ظرف تیتراسیون (سلول رسانایی) قرار می گیرند تا با محلول تیتر شده تماس نداشته باشند. بر اساس نتایج اندازه گیری، منحنی تیتراسیون هدایت سنجی ترسیم می شود. نقطه پایانی تیتراسیون با برون یابی بخش هایی از منحنی تیتراسیون در ناحیه ای که شیب آن تغییر می کند پیدا می شود. فصل 4. آنالیز هدایت سنجی (رسانایی سنجی) 4.1 ماهیت روش تجزیه و تحلیل پلاروگرافی (پلاروگرافی) مبتنی بر استفاده از روابط زیر بین پارامترهای الکتریکی یک سلول الکتروشیمیایی (در این مورد، پلاروگرافی) است که یک پتانسیل خارجی به آن اعمال می شود، و خواص محلول مورد تجزیه و تحلیل موجود در آن. الف) تجزیه و تحلیل پولاروگرافی کیفی از رابطه بین بزرگی پتانسیل الکتریکی خارجی اعمال شده بر روی میکروالکترود استفاده می کند، که در آن کاهش (یا اکسیداسیون) آنالیت بر روی میکروالکترود در شرایط معین مشاهده می شود، و ماهیت ماده در حال کاهش (یا اکسید شده). ب) در آنالیز پولاروگرافیک کمی، از رابطه بین مقدار جریان الکتریکی انتشار و غلظت ماده در حال تعیین (کاهنده یا اکسید کننده) در محلول مورد تجزیه و تحلیل استفاده می شود. پارامترهای الکتریکی - بزرگی پتانسیل الکتریکی اعمال شده و بزرگی جریان انتشار - با تجزیه و تحلیل منحنی های قطبش یا جریان-ولتاژ حاصل تعیین می شوند که به صورت گرافیکی وابستگی جریان الکتریکی در سلول قطبی را به بزرگی نشان می دهد. پتانسیل کاربردی میکروالکترود بنابراین، پلاروگرافی گاهی اوقات ولتامتری مستقیم نامیده می شود. روش کلاسیک پولاروگرافی تجزیه و تحلیل با استفاده از الکترود قطرهای جیوه در سال 1922 توسط دانشمند چک یاروسلاو هیروفسکی (1967-1890) توسعه و پیشنهاد شد، اگرچه خود الکترود قطرهای جیوه توسط فیزیکدان چکی B. Kucera در سال 1903 استفاده شد. در سال 1925. J. Heyrovsky و M. Shikata اولین پلاروگراف را طراحی کردند که امکان ثبت خودکار منحنی های قطبش را فراهم کرد. پس از آن، اصلاحات مختلف روش پلاروگرافی توسعه یافت. مقدار میانگین جریان انتشار با معادله ایلکویچ (25) تعیین می شود: که در آن K ضریب تناسب است، c غلظت (mmol/l) ماده دپلاریز کننده فعال قطبی است. iD در میکرو آمپر به عنوان اختلاف بین جریان محدود کننده و جریان باقیمانده اندازه گیری می شود. ضریب تناسب K در معادله ایلکویچ به تعدادی پارامتر بستگی دارد و برابر است با K=607nD12m23τ16 که در آن n تعداد الکترون هایی است که در واکنش ردوکس الکترود شرکت می کنند. D ضریب انتشار ماده کاهنده (cm2/s) است. t جرم جیوه ای است که در هر ثانیه از مویرگ خارج می شود (میلی گرم). t زمان تشکیل (بر حسب ثانیه) یک قطره جیوه در پتانسیل نیم موج است (معمولاً 3-5 ثانیه است). از آنجایی که ضریب انتشار D به دما بستگی دارد، ضریب تناسب K در معادله ایلکویچ با دما تغییر می کند. برای محلولهای آبی در محدوده دمایی 20 تا 50 درجه سانتیگراد، ضریب انتشار مواد دپلاریزهکننده فعال پلاروگرافیک با افزایش دما به میزان یک درجه، تقریباً 3 درصد افزایش مییابد که منجر به افزایش میانگین iD جریان انتشار تا 1 ~ میشود. -2 درصد بنابراین، پلاروگرافی در دمای ثابت انجام می شود و سلول پلاروگرافی معمولاً در دمای 0.5 ± 25 درجه سانتیگراد ترموستات می شود. جرم جیوه t و زمان تشکیل قطره t به ویژگی های الکترود ریزش جیوه و ارتفاع ستون جیوه در مویرگ و در مخزن متصل به مویین بستگی دارد. مویرگ شیشه ای میکروالکترود چکاننده جیوه معمولاً دارای قطر خارجی 3-7 میلی متر، قطر داخلی 0.03 تا 0.05 میلی متر و طول 6-15 سانتی متر است.ارتفاع ستون جیوه از انتهای پایینی مویین. تا سطح بالایی سطح جیوه در مخزن 40-80 سانتی متر است. محتوای الکترولیت بی تفاوت در محلول پلاروگراف تجزیه شده باید تقریباً 100 برابر بیشتر از محتوای ماده دپلاریز کننده تعیین شده باشد و یون های الکترولیت زمینه نباید در شرایط پلاروگرافی تخلیه شوند تا زمانی که ماده فعال پلاروگرافی تخلیه شود. پلاروگرافی با استفاده از آب، مخلوط های آلی آب (آب - اتانول، آب - استون، آب - دی متیل فرمامید و غیره) و محیط های غیر آبی (اتانول، استون، دی متیل فرمامید، دی متیل سولفوکسید و غیره) به عنوان حلال انجام می شود. قبل از شروع پلاروگرافی، جریانی از گاز بی اثر (نیتروژن، آرگون و غیره) از محلول آنالیز شده عبور می کند تا اکسیژن محلول را حذف کند، که همچنین بر اساس طرح زیر یک موج پلاروگرافی به دلیل کاهش تولید می کند: 2Н+ + 2е = Н202 Н202 + 2Н+ + 2е = 2Н20 گاهی اوقات - در مورد محلول های قلیایی - به جای عبور جریان گاز بی اثر، مقدار کمی از یک عامل احیا کننده فعال - سولفیت سدیم، متول - به محلول تجزیه شده اضافه می شود که با واکنش با آن اکسیژن محلول را متصل می کند. 4.2 تجزیه و تحلیل کمی پلاروگرافی از مطالب فوق چنین استنباط می شود که تجزیه و تحلیل کمی پلاروگرافی بر اساس اندازه گیری iD جریان انتشار به عنوان تابعی از غلظت ماده دپلاریز کننده فعال قطبی تعیین شده در محلول پلاروگرافی شده است. هنگام تجزیه و تحلیل پلاروگرام های حاصل، غلظت آنالیت با استفاده از روش های منحنی کالیبراسیون، اضافات استاندارد و محلول های استاندارد تعیین می شود. الف) روش منحنی کالیبراسیون بیشتر مورد استفاده قرار می گیرد. با استفاده از این روش، یک سری محلول استاندارد تهیه می شود که هر کدام حاوی غلظت مشخصی از آنالیت هستند. هر محلول پلاروگراف می شود (پس از دمیدن جریان گاز بی اثر از طریق آن) در شرایط یکسان، پلاروگرام ها به دست می آیند و مقادیر E12 (برای همه محلول ها یکسان) و iD جریان انتشار (برای همه محلول ها متفاوت است) پیدا می شود. . بر اساس داده های به دست آمده، یک نمودار کالیبراسیون در مختصات iD-c ساخته می شود که معمولاً یک خط مستقیم مطابق با معادله ایلکویچ است. سپس، پلاروگرافی روی محلول آنالیز شده با غلظت ناشناخته c(X) آنالیت انجام می شود، پلاروگرام به دست می آید، جریان انتشار iD(X) اندازه گیری می شود و غلظت c(X) از نمودار کالیبراسیون پیدا می شود. . ب) روش جمع استاندارد. پلاروگرام محلول آنالیز شده با غلظت ناشناخته c(X) آنالیت به دست می آید و مقدار جریان انتشار یافت می شود، یعنی. ارتفاع h پلاروگرام سپس مقدار مشخصی از آنالیت به محلول آنالیز شده اضافه می شود و غلظت آن را افزایش می دهد مقدار c(st)، پلاروگرافی دوباره انجام می شود و مقدار جدیدی از جریان انتشار پیدا می شود - ارتفاع پلاروگرام h + △ ساعت مطابق با معادله ایلکویچ (25)، می توان نوشت: h = Kc(X)، △ h = Kc(st)، جایی که ساعت △ h = c(X)c(st) و c(X) = h △ hc (st) ج) روش حل استاندارد. تحت شرایط یکسان، دو محلول پلاروگراف می شوند: یک محلول آزمایشی با غلظت ناشناخته c(X) و یک محلول استاندارد با غلظت دقیق c(st) ماده در حال تعیین. از پلاروگرام های حاصل، ارتفاع امواج پلاروگرافی h(X) و h(st) یافت می شود که به ترتیب مربوط به جریان انتشار در غلظت های c(X) و c(st) است. با توجه به معادله ایلکویچ (25) داریم: (X) = Kc(X)، h(st) = Kc(st)، محلول استاندارد طوری تهیه می شود که غلظت آن تا حد امکان به غلظت محلول مورد نظر نزدیک شود. در این شرایط، خطای تعیین به حداقل می رسد. 3 کاربردهای پلاروگرافی کاربرد روش. پلاروگرافی برای تعیین مقادیر کمی از مواد معدنی و آلی استفاده می شود. هزاران تکنیک تجزیه و تحلیل کمی پلاروگرافی توسعه داده شده است. روشهایی برای تعیین پلاروگرافی تقریباً تمام کاتیونهای فلزی، تعدادی آنیون (یونهای برمات، یدات، نیترات، پرمنگنات)، ترکیبات آلی کلاسهای مختلف حاوی گروههای دیازو، کربونیل، پراکسید، گروههای اپوکسی، پیوندهای دوگانه کربن-کربن پیشنهاد شدهاند. ، و همچنین پیوندهای کربن - هالوژن ، نیتروژن - اکسیژن ، گوگرد - گوگرد. روش فارمکوپه برای تعیین اسید سالیسیلیک، نورسولفازول، آلکالوئیدهای ویتامین B، اسید فولیک، کلین در پودر و قرص، نیکوتین آمید، پیریدوکسین هیدروکلراید، فرآوردههای آرسنیک، گلیکوزیدهای قلبی و همچنین اکسیژن و ناخالصیهای مختلف در داروسازی استفاده میشود. این روش دارای حساسیت بالایی است (تا 10 اینچ 5-10T6 mol/l)؛ گزینش پذیری؛ تکرارپذیری نسبتاً خوب نتایج (تا 2٪)؛ طیف گسترده ای از کاربردها؛ به شما امکان می دهد مخلوط مواد را بدون جداسازی آنها تجزیه و تحلیل کنید، رنگی. محلولها، حجمهای کمی از محلولها (حجم سلولهای پلاروگرافیک میتواند به کوچکی 1 میلیلیتر باشد)؛ تجزیه و تحلیل را در جریان محلول انجام دهید؛ تجزیه و تحلیل را خودکار کنید. از معایب این روش می توان به سمیت جیوه، اکسیداسیون نسبتاً آسان آن در حضور مواد اکسید کننده و پیچیدگی نسبی تجهیزات مورد استفاده اشاره کرد. انواع دیگر روش پلاروگرافی. علاوه بر پلاروگرافی کلاسیک که در بالا توضیح داده شد، که از یک میکروالکترود جیوه چکاننده با پتانسیل الکتریکی به طور یکنواخت در یک جریان الکتریکی ثابت بر روی آن افزایش می یابد، انواع دیگری از روش پلاروگرافی توسعه داده شده است - پلاروگرافی مشتق، دیفرانسیل، پالس، پلاروگرافی اسیلوگرافی. پلاروگرافی جریان متناوب - همچنین در نسخه های مختلف. فصل 5. تیتراسیون آمپرومتریک ماهیت روش. تیتراسیون آمپرومتریک (تیتراسیون پلاریزاسیون پتانسیواستاتیک) نوعی روش ولتامتری (همراه با پلاروگرافی) است. این بر اساس اندازه گیری جریان بین الکترودهای یک سلول الکتروشیمیایی است که ولتاژ خاصی به عنوان تابعی از حجم تیتر اضافه شده به آن اعمال می شود. مطابق با معادله ایلکویچ (25): ID جریان انتشار در سلول پلاروگرافی بیشتر است، هر چه غلظت c ماده فعال پلاروگرافی بیشتر باشد. اگر هنگام افزودن یک تیترانت به محلول تیتر شده تجزیه و تحلیل شده واقع در یک سلول پلاروگرافی، غلظت چنین ماده ای کاهش یا افزایش یابد، جریان انتشار نیز بر این اساس کاهش یا افزایش می یابد. نقطه هم ارزی با تغییر شدید در کاهش یا افزایش جریان انتشار تعیین می شود که مربوط به پایان واکنش ماده تیتر شده با تیتر کننده است. بین تیتراسیون آمپرومتریک با یک الکترود قابل قطبش، که تیتراسیون با محدود کردن جریان، تیتراسیون پلاروگرافی یا پلاریمتری نیز نامیده می شود، و تیتراسیون آمپرومتریک با دو الکترود قابل قطبش یکسان، یا تیتراسیون "تا زمانی که جریان به طور کامل متوقف شود"، تیتراسیون بی آمپرومتریک، تمایز قائل می شود. تیتراسیون آمپرومتریک با یک الکترود قطبش پذیر. این بر اساس اندازه گیری جریان در یک سلول پلاروگرافی بسته به مقدار تیتر اضافه شده در یک پتانسیل خارجی ثابت روی میکروالکترود، کمی بالاتر از پتانسیل نیمه موج در منحنی جریان-ولتاژ ماده تیتر شده X یا تیترانت T است. به طور معمول، پتانسیل خارجی انتخاب شده مربوط به منطقه محدود کننده جریان در پلاروگرام X یا T است. تیتراسیون بر روی یک تاسیسات متشکل از یک منبع جریان مستقیم با ولتاژ قابل تنظیم انجام می شود که یک گالوانومتر و یک سلول تیتراسیون پلاروگرافی به صورت سری به آن متصل می شوند. الکترود کار (نشان دهنده) سلول می تواند یک الکترود ریزش جیوه، یک الکترود پلاتین یا گرافیت ثابت یا دوار باشد. هنگام استفاده از الکترودهای جامد، لازم است محلول را در طول تیتراسیون هم بزنید. الکترودهای کلرید نقره یا کالومل به عنوان الکترود مرجع استفاده می شوند. پسزمینه، بسته به شرایط، الکترولیتهای مختلف قطبی غیرفعال در یک پتانسیل معین (HN03، H2S04، NH4NO3، و غیره) است. اول، منحنی های جریان-ولتاژ (پلاروگرام) برای X و T تحت شرایط یکسانی که تیتراسیون آمپرومتریک قرار است انجام شود، به دست می آید. بر اساس در نظر گرفتن این منحنی ها، یک مقدار بالقوه انتخاب می شود که در آن مقدار جریان محدود X یا T فعال قطبی به دست می آید.مقدار پتانسیل انتخاب شده در طول فرآیند تیتراسیون ثابت نگه داشته می شود. غلظت تیترانت T مورد استفاده برای تیتراسیون آمپرومتریک باید تقریباً 10 برابر بیشتر از غلظت X باشد. در این حالت عملاً نیازی به اصلاح رقت محلول در طی تیتراسیون نیست. در غیر این صورت تمام شرایط لازم برای اخذ پلاروگرام رعایت می شود. الزامات ترموستات کمتر از پلاروگرافی مستقیم است، زیرا پایان تیتراسیون نه با مقدار مطلق جریان انتشار، بلکه با تغییر شدید مقدار آن تعیین می شود. محلول آنالیز شده حاوی X به سلول پلاروگرافی اضافه می شود و تیترانت T در قسمت های کوچک اضافه می شود و هر بار جریان i اندازه گیری می شود. بزرگی جریان i به غلظت ماده فعال قطبی بستگی دارد. در نقطه هم ارزی، مقدار i به شدت تغییر می کند. بر اساس نتایج تیتراسیون آمپرومتریک، منحنی های تیتراسیون ساخته می شوند. منحنی تیتراسیون آمپرومتریک یک نمایش گرافیکی از تغییر جریان / به عنوان تابعی از حجم V تیتر اضافه شده است. منحنی تیتراسیون در مختصات جریان i - حجم V تیتر اضافه شده T (یا درجه تیتراسیون) رسم می شود. بسته به ماهیت ماده تیتر شده X و تیترانت T، منحنی های تیتراسیون آمپرومتریک می توانند انواع مختلفی داشته باشند. تیتراسیون بیامپرومتریک با هم زدن شدید محلول در مجموعه ای متشکل از یک منبع جریان مستقیم با یک پتانسیومتر انجام می شود که از آن یک اختلاف پتانسیل قابل تنظیم (0.05-0.25 V) از طریق یک میکرو آمپرمتر حساس به الکترودهای سلول الکتروشیمیایی عرضه می شود. قبل از تیتراسیون، محلولی که باید تیتر شود به محلول دوم اضافه میشود و تیترکننده در قسمتهایی اضافه میشود تا زمانی که جریان به طور ناگهانی متوقف شود یا ظاهر شود، همانطور که با خواندن یک میکرو آمپرمتر قضاوت میشود. الکترودهای پلاتین مورد استفاده در سلول الکتروشیمیایی به طور دوره ای با غوطه ور کردن آنها به مدت 30 دقیقه در اسید نیتریک غلیظ در حال جوش حاوی مواد افزودنی کلرید آهن و سپس شستشوی الکترودها با آب تمیز می شوند. تیتراسیون بیامپرومتریک یک روش دارویی است. در یدومتری، نیتریتومتری، آکوامتری و برای تیتراسیون در محیط های غیر آبی استفاده می شود. فصل 6. آنالیز کولومتری (کولومتری) 1 اصول روش کولومتری تیتراسیون هدایت سنجی الکتروشیمیایی تجزیه و تحلیل کولومتری (کولومتری) بر اساس استفاده از رابطه بین جرم m ماده ای است که در حین الکترولیز در یک سلول الکتروشیمیایی واکنش نشان می دهد و مقدار الکتریسیته Q عبور داده شده از سلول الکتروشیمیایی در طی الکترولیز فقط این ماده. بر اساس قانون واحد الکترولیز M فارادی، جرم t (بر حسب گرم) به مقدار الکتریسیته Q (بر حسب کولن) با رابطه (27) مرتبط است. که در آن M جرم مولی ماده ای است که در طول الکترولیز واکنش داده است، g/mol. n تعداد الکترون های شرکت کننده در واکنش الکترود است. 96487 C/mol عدد فارادی است. مقدار الکتریسیته Q (بر حسب C) که در طول الکترولیز از یک سلول الکتروشیمیایی عبور می کند برابر است با حاصلضرب جریان الکتریکی i (در A) و زمان الکترولیز. τ ( در ج): اگر مقدار الکتریسیته Q اندازهگیری شود، طبق (27) جرم m را میتوان محاسبه کرد. ماده داده شده؛ فرآیندهای جانبی باید حذف شوند. به عبارت دیگر، خروجی جریان (بازده) باید 100٪ باشد. از آنجایی که مطابق قانون یکپارچه الکترولیز M. Faraday، برای تعیین جرم t (g) ماده ای که در طول الکترولیز واکنش می دهد، لازم است مقدار الکتریسیته Q صرف شده برای تبدیل الکتروشیمیایی ماده در حال تعیین اندازه گیری شود. ، در کولن به روش کولومتری می گویند. وظیفه اصلی اندازه گیری های کولومتریک تعیین مقدار الکتریسیته Q تا حد امکان دقیق است. تجزیه و تحلیل کولومتریک یا در حالت آمپروستاتیک (گالوانوستاتیک) انجام می شود، یعنی. با جریان الکتریکی ثابت i=const، یا با پتانسیل ثابت کنترل شده الکترود کار (کولومتری پتانسیواستاتیک)، زمانی که جریان الکتریکی در طول فرآیند الکترولیز تغییر می کند (کاهش می یابد). در حالت اول برای تعیین مقدار الکتریسیته Q کافی است زمان الکترولیز t(s)، جریان مستقیم /(A) را تا حد امکان دقیق اندازه گیری و مقدار Q را با استفاده از فرمول (10.28) محاسبه کرد. در حالت دوم، مقدار Q یا با محاسبه یا با استفاده از کولومترهای شیمیایی تعیین می شود. کولومتری مستقیم و کولومتری غیرمستقیم (تیتراسیون کولومتری) وجود دارد. ماهیت روش. کولومتری مستقیم در جریان ثابت به ندرت استفاده می شود. بیشتر اوقات، کولومتری با پتانسیل ثابت کنترل شده الکترود کار یا کولومتری پتانسیواستاتیک مستقیم استفاده می شود. در کولومتری پتانسیواستاتیک مستقیم، ماده ای که تعیین می شود مستقیماً الکترولیز می شود. مقدار برق مصرف شده برای الکترولیز این ماده اندازه گیری می شود و جرم m ماده در حال تعیین با استفاده از معادله محاسبه می شود. در طی فرآیند الکترولیز، پتانسیل الکترود کار ثابت نگه داشته می شود، E = const، که معمولاً از دستگاه ها - پتانسیواستات ها - استفاده می شود. مقدار ثابت پتانسیل E از قبل بر اساس در نظر گرفتن منحنی جریان-ولتاژ (پلاریزاسیون) ساخته شده در مختصات جریان i - پتانسیل E (همانطور که در پلاروگرافی انجام می شود) انتخاب می شود، که در شرایط مشابهی که الکترولیز به دست می آید. انجام شد. به طور معمول، یک مقدار پتانسیل E انتخاب می شود که مربوط به ناحیه جریان محدود کننده برای ماده مورد تجزیه و تحلیل است و کمی بالاتر از پتانسیل نیمه موج E12 آن (با -0.05-0.2 V) است. در این مقدار پتانسیل، مانند پلاروگرافی، الکترولیت زمینه نباید تحت الکترولیز قرار گیرد. همانطور که فرآیند الکترولیز در یک پتانسیل ثابت پیش می رود، جریان الکتریکی در سلول کاهش می یابد، زیرا غلظت ماده الکتریکی شرکت کننده در واکنش الکترود کاهش می یابد. در این حالت، جریان الکتریکی در طول زمان طبق یک قانون نمایی از مقدار اولیه i0 در زمان t = O به مقدار i در زمان t کاهش می یابد: که در آن ضریب k به ماهیت واکنش، هندسه سلول الکتروشیمیایی، مساحت الکترود کار، ضریب انتشار ماده تعیین شده، سرعت هم زدن محلول و حجم آن بستگی دارد. روشهای تعیین مقدار برق عبوری از محلول در کولومتری پتانسیواستاتیک مستقیم. مقدار Q را می توان با روش های محاسباتی یا با استفاده از یک کولومتر شیمیایی تعیین کرد. الف) محاسبه کمیت Q از ناحیه زیر منحنی i در مقابل m. برای تعیین Q بدون خطای محسوس، روش نیاز به تکمیل تقریباً کامل فرآیند الکترولیز دارد. برای مدت طولانی در عمل، همانطور که در بالا ذکر شد، مساحت با مقدار m مربوطه اندازه گیری می شود 0.001i0 (0.1٪ از i0). ب) محاسبه مقدار Q بر اساس وابستگی In / به m بر اساس آن داریم: Q = 0∞i0e-k τ د τ =i00∞e-k τ د τ =i0k از آنجا که ∞i0e-k τ د τ = - k-1 e-k∞-e-k0= k-10-1=k-1 کاربرد کولومتری مستقیم این روش دارای گزینش پذیری بالا، حساسیت (تا 10~8-10~9 گرم یا تا ~10~5 mol/l)، قابلیت تکرارپذیری (تا ~1-2٪) است و امکان تعیین محتوای ریز ناخالصی ها را فراهم می کند. از معایب این روش می توان به شدت کار بالا و مدت زمان آنالیز و نیاز به تجهیزات گران قیمت اشاره کرد. کولومتری مستقیم می تواند برای تعیین - در طی احیای کاتدی - یون های فلزی، مشتقات آلی نیترو و هالوژن استفاده شود. در طی اکسیداسیون آندی - آنیون های کلرید، برمید، یدید، تیوسیانات، یون های فلزی در حالت های اکسیداسیون پایین تر هنگام تبدیل آنها به حالت های اکسیداسیون بالاتر، به عنوان مثال: As(IH) -> As(V)،Cr(II) -> Cr(III) )، Fe(II) -» Fe(III)، T1(I) -> Tl(III)، و غیره. در آنالیزهای دارویی از کولومتری مستقیم برای تعیین اسیدهای اسکوربیک و پیکریک، نووکائین، هیدروکسی کینولین و در برخی موارد دیگر استفاده می شود. همانطور که در بالا ذکر شد، کولومتری مستقیم کاملاً کار فشرده و زمان بر است. علاوه بر این، در برخی موارد، فرآیندهای جانبی حتی قبل از اتمام واکنش الکتروشیمیایی اصلی به طور قابل توجهی شروع میشوند که بازده جریان را کاهش میدهد و میتواند منجر به خطاهای تجزیه و تحلیل قابل توجهی شود. بنابراین کولومتری غیرمستقیم - تیتراسیون کولومتریک - بیشتر مورد استفاده قرار می گیرد. 3 تیتراسیون کولومتریک ماهیت روش. در تیتراسیون کولومتری، آنالیت X که در محلول در یک سلول الکتروشیمیایی قرار دارد، با "تیترانت" T واکنش می دهد، ماده ای که به طور مداوم در الکترود ژنراتور در طول الکترولیز یک ماده کمکی موجود در محلول تشکیل می شود (تولید می شود). پایان تیتراسیون - لحظه ای که تمام آنالیت X به طور کامل با "تیترانت" T تولید شده واکنش نشان می دهد، یا به صورت بصری با روش نشانگر، وارد محلول یک نشانگر مناسب که در نزدیکی TE تغییر رنگ می دهد، یا با استفاده از روش های ابزاری ثبت می شود. - از نظر پتانسیومتری، آمپرومتری، نورسنجی. بنابراین، در تیتراسیون کولومتری، تیتر از بورت به محلول در حال تیتر اضافه نمی شود. نقش تیترانت توسط ماده T ایفا می شود که به طور مداوم در طی واکنش الکترود روی الکترود ژنراتور تولید می شود. بدیهی است که بین تیتراسیون معمولی، زمانی که تیتراتور از خارج وارد محلول تیتر شده می شود و با افزودن آن با آنالیت واکنش می دهد و تولید ماده T که با تشکیل آن نیز واکنش نشان می دهد، قیاس وجود دارد. با آنالیت بنابراین، روش مورد بررسی، تیتراسیون کولومتریک نامیده می شود. تیتراسیون کولومتریک در حالت آمپروستاتیک (گالوانوستاتیک) یا پتانسیواستاتیک انجام می شود. بیشتر اوقات، تیتراسیون کولومتریک در حالت آمپروستاتیک انجام می شود و جریان الکتریکی را در کل زمان الکترولیز ثابت نگه می دارد. در تیتراسیون کولومتری به جای حجم تیترانت اضافه شده، زمان t و جریان i الکترولیز اندازه گیری می شود. فرآیند تشکیل ماده T در سلول کولومتریک در طی الکترولیز تولید تیترانت نامیده می شود. تیتراسیون کولومتریک در جریان ثابت. در طول تیتراسیون کولومتری در حالت آمپروستاتیک (در جریان ثابت)، زمان t که در طی آن الکترولیز انجام شده است، اندازه گیری می شود و مقدار الکتریسیته مصرف شده در طول الکترولیز با استفاده از فرمول محاسبه می شود، پس از آن جرم آنالیت X با استفاده از آن بدست می آید. نسبت. بنابراین، برای مثال، استانداردسازی محلول اسید هیدروکلریک HC1 با روش تیتراسیون کولومتریک با تیتر کردن یون های هیدروژن H30+ از محلول استاندارد شده حاوی HC1 انجام می شود که به طور الکتریکی در کاتد پلاتین توسط یون های هیدروکسید OH- در طی الکترولیز تولید می شود. اب: Н20 + 2е = 20Н- + Н2 تیترانت حاصل - یون هیدروکسید - با یون های H30+ در محلول واکنش می دهد: H30+ + OH- = 2H20 تیتراسیون در حضور نشانگر فنل فتالئین انجام می شود و با ظاهر شدن رنگ صورتی روشن محلول متوقف می شود. با دانستن مقدار جریان مستقیم i (بر حسب آمپر) و زمان t (بر حسب ثانیه) که برای تیتراسیون صرف شده است، مقدار الکتریسیته Q (بر حسب کولن) با استفاده از فرمول (28) و جرم (بر حسب گرم) HC1 واکنش نشان داده شده محاسبه می شود. در فرمول (27) در قسمتی از محلول استاندارد شده HC1 به سلول کولومتری (به ظرف ژنراتور) اضافه شده است. شرایط تیتراسیون کولومتریک از موارد فوق چنین استنباط می شود که شرایط انجام تیتراسیون کولومتریک باید 100% راندمان جریان را تضمین کند. برای انجام این کار، شما باید حداقل شرایط زیر را برآورده کنید. الف) معرف کمکی که از آن تیترانت در الکترود کار تولید می شود باید در محلول به مقدار زیادی نسبت به ماده تعیین شده (1000 برابر بیش از حد) موجود باشد. تحت این شرایط، واکنش های الکتروشیمیایی جانبی معمولا حذف می شوند، که اصلی ترین آنها اکسیداسیون یا کاهش الکترولیت زمینه است، به عنوان مثال، یون های هیدروژن: Н+ + 2е = Н2 ب) مقدار جریان مستقیم i=const در طول الکترولیز باید کمتر از مقدار جریان انتشار معرف کمکی باشد تا از واکنشی که شامل یونهای الکترولیت زمینه است جلوگیری شود. ج) لازم است تا حد امکان مقدار برق مصرفی در حین الکترولیز به دقت تعیین شود که مستلزم ثبت دقیق شروع و پایان شمارش زمان و مقدار جریان الکترولیز است. تیتراسیون کولومتریک در پتانسیل ثابت. حالت پتانسیواستاتیک کمتر در تیتراسیون کولومتری استفاده می شود. تیتراسیون کولومتریک در حالت پتانسیواستاتیک با یک مقدار پتانسیل ثابت مطابق با پتانسیل تخلیه یک ماده روی الکترود کار انجام می شود، به عنوان مثال، در حین احیای کاتدی کاتیون های فلزی M "* روی یک الکترود کاری پلاتین. به عنوان واکنش. در ادامه، پتانسیل ثابت می ماند تا زمانی که همه کاتیون های فلزی واکنش نشان دهند، پس از آن به شدت کاهش می یابد، زیرا دیگر کاتیون های فلزی تعیین کننده پتانسیل در محلول وجود ندارد. کاربرد تیتراسیون کولومتریک. در تیتراسیون کولومتریک، می توان از انواع واکنش های آنالیز تیتریمتری استفاده کرد: واکنش های اسید-باز، ردوکس، رسوب، واکنش های کمپلکس. بنابراین، مقادیر کمی از اسیدها را می توان با تیتراسیون اسید-باز کولومتریک با یون های OH- تولید شده در طول الکترولیز آب در کاتد تعیین کرد: Н20 + 2е = 20Н" + Н2 همچنین می توان پایه ها را با یون های هیدروژن H+ تولید شده در آند در طی الکترولیز آب تیتر کرد: Н20-4е = 4Н+ + 02 با تیتراسیون کولومتری برومومتری ردوکس، می توان ترکیبات آرسنیک (III)، آنتیموان (III)، یدیدها، هیدرازین، فنل ها و سایر مواد آلی را تعیین کرد. برم که به صورت الکتریکی در آند تولید می شود، به عنوان یک تیتر کننده عمل می کند: VG -2e = Vg2 تیتراسیون کولومتریک رسوبی میتواند یونهای هالید و ترکیبات حاوی گوگرد آلی را توسط کاتیونهای نقره Ag+، کاتیونهای روی Zn2+ توسط یونهای فروسیانید الکتروتولید شده و غیره تعیین کند. تیتراسیون کولومتریک کمپلکسومتری کاتیون های فلزی را می توان با آنیون های EDTA که بر روی یک کاتد کمپلکسونات جیوه (I) تولید می شوند انجام داد. تیتراسیون کولومتریک دارای دقت بالایی است، طیف گسترده ای از کاربردها در تجزیه و تحلیل کمی، به شما امکان می دهد مقادیر کمی از مواد، ترکیبات کم مقاومت را تعیین کنید (زیرا آنها بلافاصله پس از تشکیل واکنش نشان می دهند)، به عنوان مثال، مس (1)، نقره (H) قلع (P)، تیتانیوم (III)، منگنز (III)، کلر، برم و غیره. مزایای روش همچنین شامل این واقعیت است که آماده سازی، استانداردسازی و ذخیره سازی تیترانت مورد نیاز نیست، زیرا به طور مداوم در طول الکترولیز تشکیل می شود و بلافاصله در واکنش با ماده در حال تعیین مصرف می شود. نتیجه روش های الکتروشیمیایی آنالیز بر اساس فرآیندهایی است که روی الکترودها یا فضای بین الکترودها اتفاق می افتد. روش های الکتروشیمیایی آنالیز یکی از قدیمی ترین روش های فیزیکوشیمیایی آنالیز است (برخی در اواخر قرن نوزدهم شرح داده شدند). مزیت آنها دقت بالا و سادگی نسبی تجهیزات و تکنیک های تجزیه و تحلیل است. دقت بالا توسط قوانین بسیار دقیق مورد استفاده در روش های الکتروشیمیایی تجزیه و تحلیل، به عنوان مثال، قانون فارادی تعیین می شود. یک راحتی بزرگ این است که آنها از تأثیرات الکتریکی استفاده می کنند و این واقعیت است که نتیجه این تأثیر (پاسخ) در قالب یک سیگنال الکتریکی به دست می آید. این امر سرعت و دقت بالای خواندن را تضمین می کند و امکانات گسترده ای را برای اتوماسیون باز می کند. روش های الکتروشیمیایی آنالیز با حساسیت و گزینش پذیری خوب متمایز می شوند، در برخی موارد می توان آنها را به عنوان میکروآنالیز طبقه بندی کرد، زیرا گاهی اوقات کمتر از 1 میلی لیتر محلول برای تجزیه و تحلیل کافی است. ابزار آنها یک سلول الکتروشیمیایی است که یک ظرف با محلول الکترولیت است که حداقل دو الکترود در آن غوطه ور است. بسته به مشکل حل شده، شکل و جنس ظرف، تعداد و ماهیت الکترودها، محلول و شرایط تجزیه و تحلیل (ولتاژ اعمال شده (جریان) و سیگنال تحلیلی ثبت شده، دما، هم زدن، تصفیه با گاز بی اثر و غیره. ) ممکن است متفاوت باشد. ماده ای که تعیین می شود می تواند هم بخشی از الکترولیت پرکننده سلول و هم یکی از الکترودها باشد. روش های تجزیه و تحلیل الکتروشیمیایی نقش مهمی در دنیای مدرن ایفا می کنند. در زمان ما مراقبت از محیط زیست از اهمیت ویژه ای برخوردار است. با استفاده از این روش ها می توان محتوای تعداد زیادی از مواد آلی و معدنی مختلف را تعیین کرد. آنها اکنون در شناسایی مواد خطرناک موثرتر هستند.
ارسال کار خوب خود در پایگاه دانش ساده است. از فرم زیر استفاده کنید
دانشجویان، دانشجویان تحصیلات تکمیلی، دانشمندان جوانی که از دانش پایه در تحصیل و کار خود استفاده می کنند از شما بسیار سپاسگزار خواهند بود.
نوشته شده در http://www.allbest.ru
وزارت آموزش و پرورش و علوم فدراسیون روسیه
موسسه آموزشی بودجه ایالتی فدرال
آموزش عالی
"دانشگاه فنی تحقیقات ملی ایرکوتسک"
گروه متالورژی فلزات غیرآهنی
(نام بخش)
"روش تحقیق الکتروشیمیایی"
چکیده در مورد رشته
"روش های فیزیکوشیمیایی برای مطالعه فرآیندهای متالورژی"
توسط دانش آموزی از گروه MCM-16-1 تکمیل شد
زاخارنکوف آر.آی.
توسط معلم بخش MCM بررسی شد
کوزمینا ام.یو.
ایرکوتسک 2017
معرفی
الکتروشیمی شاخهای از شیمی فیزیک است که سیستمهای حاوی یونها (محلول یا مذاب الکترولیتها) و فرآیندهایی را که در مرز دو فاز با مشارکت ذرات باردار اتفاق میافتند، در نظر میگیرد.
اولین ایده ها در مورد رابطه بین پدیده های شیمیایی و الکتریکی در قرن 18 شناخته شد، زیرا تعداد زیادی آزمایش فیزیکی و شیمیایی با تخلیه الکتریکی و رعد و برق، با بارهای واقع در شیشه های لیدن انجام شد، اما همه آنها تصادفی بودند. طبیعت به دلیل عدم وجود منبع قوی ثابت انرژی الکتریکی. منشا الکتروشیمی با نام های L. Galvani و A. Volta مرتبط است. گالوانی هنگام مطالعه عملکردهای فیزیولوژیکی قورباغه به طور تصادفی یک مدار الکتروشیمیایی ایجاد کرد. از دو فلز مختلف و یک پای قورباغه آماده تشکیل شده بود. پنجه هم الکترولیت بود و هم نشانگر جریان الکتریکی، اما نتیجه گیری اشتباه داده شد، یعنی به گفته گالوانی، این جریان الکتریکی که در مدار بوجود آمد منشا حیوانی داشت، یعنی با ویژگی های عملکردی آن مرتبط بود. بدن قورباغه (تئوری "الکتریسیته حیوانی").
تفسیر صحیح آزمایش های گالوانی توسط A. Volta ارائه شد. او اولین باتری سلول های گالوانیکی را ایجاد کرد - یک ستون ولتایی. سلول های باتری از دیسک های مسی و روی تشکیل شده بود و الکترولیت یک ماده اسفنجی بود که در آب نمک یا اسید خیس شده بود. این اتصال بود که امکان به دست آوردن جریان الکتریکی را فراهم کرد. به زودی، از طریق آثار دانشمندان بزرگ A. Volta، J. Daniel، B. S. Jacobi، P. R. Bagration، G. Plante و دیگران، سلول ها و باتری های گالوانیکی قدرتمند، آسان برای استفاده ظاهر شدند. سپس A. Volta یک سری ولتاژ فلزی را توسعه داد. اگر دو فلز مختلف با هم تماس پیدا کرده و سپس از هم جدا شوند، با استفاده از وسایل فیزیکی مانند الکتروسکوپ می توان دریافت که یک فلز بار مثبت و دیگری بار منفی پیدا کرده است. این سری از فلزات که در آن هر فلز قبلی بار مثبت دارد، اما پس از تماس با هر فلز بعدی، یعنی سری ولتا، شبیه به سری ولتاژ است.
علاوه بر این، در آغاز قرن 19، الکترولیز توسعه یافت، و M. Faraday قوانین کمی الکترولیز را ایجاد کرد. دانشمندان کمک زیادی به توسعه الکتروشیمی کردند: S. A. Arrhenius، V. F. Ostwald، R. A. Colley، P. Debye، W. Nernst، G. Helmholtz و غیره. اکنون الکتروشیمی به نظری و کاربردی تقسیم می شود. از طریق استفاده از روش های الکتروشیمیایی با سایر شاخه های شیمی فیزیک و همچنین با شیمی تجزیه و سایر علوم مرتبط می شود.
کولومتری هدایت سنجی پتانسیومتری الکتروشیمیایی
1 . روشهای تحقیق الکتروشیمیایی
نیاز به استفاده از روش های متنوع برای مطالعه فرآیندهای الکتروشیمیایی به دلیل دامنه وسیع تغییرات در سرعت انتقال الکترون در واکنش های الکترودی است. هر یک از روش ها دارای یک حد معین در مقدار تعیین شده چگالی جریان مبادله ای است که بالاتر از آن نمی توان پارامترهای الکتروشیمیایی واکنش الکترود را تعیین کرد. در رابطه با هر شی خاص، لازم است روشی انتخاب شود که حداکثر اطلاعات قابل اعتماد را ارائه دهد. هنگام انجام مطالعات الکتروشیمیایی، شناخت ترکیب شیمیایی مواد اولیه و محصولات واکنش ضروری است. برای تعیین ترکیب الکترولیت از روش های مختلف فیزیکی و شیمیایی استفاده می شود: اسپکتروفتومتری، پتانسیومتری، تحلیلی و غیره. هنگام انجام مطالعات الکتروشیمیایی، شرایط زیر باید رعایت شود.
1. حداکثر خلوص معرف های مورد استفاده. ترکیب الکترودها و همچنین وضعیت سطوح آنها باید کاملاً مشخص باشد. باید دقت شود که سطح الکترودها در طول فرآیند اندازه گیری دچار تغییر نشود.
2. طراحی سلول الکتروشیمیایی و الکترودهای واقع در آن باید توزیع یکنواخت جریان را در کل سطح الکترود کار تضمین کند.
3. اندازه گیری ها باید در دماهای کاملاً کنترل شده انجام شود.
4. فشار و ترکیب فاز گاز بالای الکترولیت را ثابت نگه دارید. به عنوان یک قاعده، مطالعات در یک محیط گاز بی اثر (N 2، Ar، Ne، He H 2) انجام می شود، زیرا اکسیژن در فاز گاز می تواند تأثیر قابل توجهی بر مکانیسم فرآیند داشته باشد.
5. لازم است از چنین شرایط آزمایشی اطمینان حاصل شود که تحت آن افت پتانسیل در قسمت منتشر لایه دوگانه الکتریکی حداقل یا دقیقاً شناخته شده باشد. برای کاهش این پتانسیل، به عنوان یک قاعده، از یک الکترولیت پس زمینه استفاده می شود که غلظت آن نباید کمتر از 20 برابر بیشتر از ماده اصلی باشد. با این حال، ابتدا باید مطمئن شوید که الکترولیت پس زمینه منحنی پلاریزاسیون واکنش مورد مطالعه را تحریف نمی کند.
6. اندازه گیری دقیق پتانسیل الکترود کار. برای این کار لازم است پتانسیل انتشار بین الکترولیت مورد مطالعه و الکترولیت الکترود مرجع حذف شود. این پتانسیل در هنگام نزدیک شدن به جریان محدود کننده حداکثر مقدار خود را می گیرد و می تواند نتایج اندازه گیری را به طور قابل توجهی مخدوش کند. برای از بین بردن پتانسیل انتشار بین الکترولیت مورد مطالعه و الکترولیت الکترود مرجع، مطلوب است: الف) الکترود مرجعی را انتخاب کنید که ترکیب الکترولیت مشابه مورد مطالعه را داشته باشد. به عنوان مثال، هنگام تحقیق در محلول های کلرید، استفاده از الکترودهای کلرید نقره، کلومل و کلر راحت است. در محلول های سولفات اسیدی - الکترودهای سولفات جیوه و غیره. ب) از یک الکترود مرجع با یک الکترولیت استفاده کنید که در مرز آن با الکترولیت مورد مطالعه، پتانسیل انتشار با استفاده از فرمول های شناخته شده قابل محاسبه است.
هنگام اندازه گیری در محلول هایی با قدرت یونی ثابت، و در غلظت های پس زمینه بالا - با غلظت یونی ثابت، در اصل می توانید از هر الکترود مرجع استفاده کنید. پتانسیل انتشار در این مورد می تواند بسیار بزرگ، اما همچنین ثابت باشد - می توان آن را به صورت تجربی محاسبه یا تعیین کرد.
در تمام موارد مطالعه سینتیک فرآیندهای الکتروشیمیایی، اندازه گیری چگالی جریان ضروری است. معمولاً آنها با یافتن روشهای شیمی تحلیلی و کولومتری شروع میکنند تا تعیین کنند که آیا فقط یک واکنش مورد مطالعه در الکترود رخ میدهد یا اینکه توسط واکنشهای جانبی پیچیده است. در مورد واکنش های جانبی، باید مشخص شود که چه نسبتی از جریان فقط به دلیل اجرای واکنش مورد مطالعه است (به اصطلاح مشخصه قطبش جزئی را برای واکنش مورد مطالعه بسازید).
مکانیسم واکنش الکترود را می توان تنها در صورتی تفسیر کرد که ماده اولیه به یک محصول با راندمان جریان 100٪ تبدیل شود. بررسی واکنش برای مطابقت با قانون فارادی یا انجام اندازهگیریهای کولومتری به شما امکان میدهد همزمان تعداد الکترونهای شرکتکننده در واکنش الکترود کل را تعیین کنید. دانستن ترکیب ماده اولیه و محصول واکنش و همچنین تعداد کل الکترون های منتقل شده، نوشتن معادله واکنش الکترود کل را ممکن می سازد.
مرحله بعدی در مطالعه مکانیسم واکنش الکترود این است که بفهمیم کدام مرحله محدود کننده است.
اگر مرحله محدود کننده مرحله تخلیه-یونیزاسیون باشد و بقیه به صورت برگشت پذیر پیش بروند، می توان پارامترهای جنبشی اصلی فرآیند را به صورت گرافیکی یا تحلیلی با اعمال معادلات تئوری تخلیه آهسته برای ویژگی های پلاریزاسیون تعیین کرد.
1.1 روشهای تجزیه و تحلیل الکتروشیمیایی
روش های الکتروشیمیاییتحلیل و بررسیمجموعه ای از روش های تجزیه و تحلیل کمی و کیفی بر اساس پدیده های الکتروشیمیایی است که در محیط مورد مطالعه یا در سطح مشترک رخ می دهد و با تغییرات در ساختار، ترکیب شیمیایی یا غلظت ماده مورد تجزیه و تحلیل مرتبط است.
روش های الکتروشیمیایی مستقیم و غیر مستقیم وجود دارد. روش های مستقیم از وابستگی قدرت جریان (پتانسیل و غیره) به غلظت جزء در حال تعیین استفاده می کنند. در روشهای غیرمستقیم، قدرت جریان (پتانسیل و غیره) اندازهگیری میشود تا نقطه پایانی تیتراسیون آنالیت با یک تیترانت مناسب، یعنی. از وابستگی پارامتر اندازه گیری شده به حجم تیترانت استفاده می شود.
برای هر نوع اندازه گیری الکتروشیمیایی، یک مدار الکتروشیمیایی یا سلول الکتروشیمیایی مورد نیاز است که محلول آنالیز شده جزء لاینفک آن است.
روش های الکتروشیمیایی بسته به نوع پدیده های اندازه گیری شده در طول فرآیند آنالیز طبقه بندی می شوند. دو گروه از روش های الکتروشیمیایی وجود دارد:
1. روشهایی بدون تحمیل پتانسیل اضافی، بر اساس اندازهگیری اختلاف پتانسیلی که در یک سلول الکتروشیمیایی متشکل از یک الکترود و یک ظرف با محلول آزمایشی رخ میدهد. این گروه از روش ها نامیده می شود پتانسیومتری روشهای پتانسیومتری از وابستگی پتانسیل تعادل الکترودها به غلظت یونهای شرکتکننده در واکنش الکتروشیمیایی روی الکترودها استفاده میکنند.
2. روشهای تحمیل پتانسیل خارجی بر اساس اندازهگیری:
الف) رسانایی الکتریکی محلول ها؟ هدایت سنجی;
ب) مقدار برق عبوری از محلول؟ کولومتری;
ج) وابستگی جریان به پتانسیل اعمال شده؟ ولت آمپرومتری;
د) زمان لازم برای انجام واکنش الکتروشیمیایی - روش های کرونوالکتروشیمیایی(کرنوولتامتری، کرونو رساناسنجی).
در روش های این گروه، یک پتانسیل خارجی به الکترودهای سلول الکتروشیمیایی اعمال می شود.
عنصر اصلی ابزار برای تجزیه و تحلیل الکتروشیمیایی، سلول الکتروشیمیایی است. در روش های بدون تحمیل پتانسیل اضافی، اینطور است سلول گالوانیکی، که در آن یک جریان الکتریکی به دلیل واکنش های ردوکس شیمیایی رخ می دهد. در سلولی مانند سلول گالوانیکی، دو الکترود با محلول مورد تجزیه و تحلیل در تماس هستند - یک الکترود نشانگر، که پتانسیل آن به غلظت ماده بستگی دارد، و یک الکترود با پتانسیل ثابت - یک الکترود مرجع، که در برابر آن پتانسیل الکترود نشانگر اندازه گیری می شود. تفاوت پتانسیل با استفاده از دستگاه های ویژه - پتانسیومتر اندازه گیری می شود.
در روش هایی با تحمیل پتانسیل خارجی، سلول الکتروشیمیاییبه این دلیل نامگذاری شده است که در الکترودهای سلول، تحت تأثیر پتانسیل اعمال شده، الکترولیز اتفاق می افتد - اکسیداسیون یا کاهش یک ماده. در آنالیز هدایت سنجی، از سلول رسانایی سنجی استفاده می شود که در آن رسانایی الکتریکی محلول اندازه گیری می شود. با توجه به روش کاربرد، روشهای الکتروشیمیایی را میتوان به روشهای مستقیم طبقهبندی کرد که در آن غلظت مواد با توجه به قرائتهای دستگاه اندازهگیری میشود و تیتراسیون الکتروشیمیایی که در آن نشاندهنده نقطه هم ارزی با استفاده از اندازهگیریهای الکتروشیمیایی ثبت میشود. مطابق با این طبقه بندی، تیتراسیون پتانسیومتری و پتانسیومتری، تیتراسیون هدایت سنجی و هدایت سنجی و غیره متمایز می شوند.
ابزارهای تعیین الکتروشیمیایی، علاوه بر سلول الکتروشیمیایی، همزن و مقاومت بار، شامل دستگاه هایی برای اندازه گیری اختلاف پتانسیل، جریان، مقاومت محلول و مقدار الکتریسیته می باشد. این اندازه گیری ها را می توان با ابزارهای اشاره گر (ولت متر یا میکرو آمپرمتر)، اسیلوسکوپ ها و پتانسیومترهای ثبت خودکار انجام داد. اگر سیگنال الکتریکی از سلول بسیار ضعیف باشد، با استفاده از تقویت کننده های رادیویی تقویت می شود. در دستگاههای روشهای با تحمیل پتانسیل خارجی، بخش مهمی از دستگاه تأمین پتانسیل مناسب جریان مستقیم یا متناوب تثبیت شده (بسته به نوع روش) به سلول است. واحد منبع تغذیه دستگاه های آنالیز الکتروشیمیایی معمولاً شامل یکسو کننده و تثبیت کننده ولتاژ است که عملکرد ثابت دستگاه را تضمین می کند.
1.2 پتانسیومتری
پتانسیومتری مبتنی بر اندازهگیری اختلاف پتانسیلهای الکتریکی است که بین الکترودهای غیرمشابه غوطهور در محلولی با ماده در حال تعیین ایجاد میشود. هنگامی که یک واکنش ردوکس (الکتروشیمیایی) روی الکترودها رخ می دهد، پتانسیل الکتریکی در الکترودها ایجاد می شود. واکنشهای ردوکس بین یک عامل اکسید کننده و یک عامل کاهنده با تشکیل جفتهای ردوکس اتفاق میافتد که پتانسیل E آن توسط معادله نرنست با غلظت اجزای جفت [ok] و [rec] تعیین میشود:
جایی که E°- پتانسیل الکترود استاندارد، V;
n- تعداد الکترون های شرکت کننده در فرآیند.
اندازه گیری های پتانسیومتری با پایین آوردن دو الکترود در محلول انجام می شود - یک الکترود نشانگر که به غلظت یون های تعیین شده واکنش نشان می دهد و یک الکترود استاندارد یا مرجع که پتانسیل نشانگر در برابر آن اندازه گیری می شود. چندین نوع نشانگر و الکترود استاندارد استفاده می شود.
الکترود از نوع اولقابل برگشت با توجه به یون های فلزی که الکترود از آن تشکیل شده است. هنگامی که چنین الکترودی به محلولی حاوی کاتیون های فلزی پایین می آید، یک جفت الکترود تشکیل می شود: م n + /م.
الکترودهای نوع دوم به آنیونها حساس هستند و فلز M با لایهای از نمک نامحلول MA با آنیون پوشیده شدهاند آ- که الکترود به آن حساس است. هنگامی که چنین الکترودی با محلولی حاوی آنیون مشخص شده تماس پیدا می کند آ-، پتانسیل E بوجود می آید که مقدار آن به محصول حلالیت نمک بستگی دارد
و غیره M.A. و غلظت آنیون [ آ-] در محلول.
الکترودهای نوع دوم عبارتند از کلرید نقره و کالومل. الکترودهای نقره اشباع کلرید و کلومل پتانسیل ثابتی را حفظ می کنند و به عنوان الکترودهای مرجع که پتانسیل الکترود نشانگر را بر اساس آن اندازه گیری می کنند، استفاده می شود.
الکترودهای بی اثر- صفحه یا سیم ساخته شده از فلزات سخت اکسیداسیون - پلاتین، طلا، پالادیوم. آنها برای اندازه گیری E در محلول های حاوی یک زوج ردوکس (به عنوان مثال Fe 3 + / Fe 2 +) استفاده می شوند.
الکترودهای غشایی انواع مختلف غشایی دارند که پتانسیل غشایی E بر روی آن پدید می آید.مقدار E بستگی به تفاوت غلظت همان یون در طرف های مختلف غشا دارد. ساده ترین و پرکاربردترین الکترود غشایی، الکترود شیشه ای است.
اختلاط نوع نمک های نامحلول AgBr، AgCl، AgIو دیگران با مقداری پلاستیک (لاستیک، پلی اتیلن، پلی استایرن) منجر به ایجاد آن شدند الکترودهای انتخابی یونیبر برادر-, Cl-, من- جذب انتخابی یون های مشخص شده از محلول به دلیل قانون Paneth - Faience - Hahn. از آنجایی که غلظت یون های قابل تشخیص در خارج از الکترود با غلظت داخل الکترود متفاوت است، تعادل روی سطوح غشا متفاوت است که منجر به ظهور پتانسیل غشایی می شود.
برای انجام تعیینهای پتانسیومتری، یک سلول الکتروشیمیایی از یک الکترود مرجع نشانگر مونتاژ میشود که در محلول مورد تجزیه و تحلیل غوطهور شده و به یک پتانسیومتر متصل میشود. الکترودهای مورد استفاده در پتانسیومتری مقاومت داخلی بالایی دارند (500-1000 MOhm)، بنابراین انواع پتانسیومترهایی وجود دارند که ولت مترهای پیچیده الکترونیکی با مقاومت بالا هستند. برای اندازه گیری EMF سیستم الکترود در پتانسیومتر، از مدار جبرانی برای کاهش جریان در مدار سلول استفاده می شود.
اغلب از پتانسیومترها برای اندازه گیری مستقیم pH، نشانگر غلظت یون های دیگر استفاده می شود. pNa، pK، pNH؟، pClو mV اندازه گیری ها با استفاده از الکترودهای انتخاب کننده یون مناسب انجام می شود.
برای اندازه گیری pH از یک الکترود شیشه ای و یک الکترود مرجع - کلرید نقره - استفاده می شود. قبل از انجام آنالیزها، لازم است کالیبراسیون pH مترها را با استفاده از محلول های بافر استاندارد که تثبیت آن به دستگاه متصل است، بررسی کنید.
PH متر علاوه بر تعیین مستقیم pH، pNa، pK، pNH؟، pClو دیگران اجازه می دهند که تیتراسیون پتانسیومتری یون تعیین شود.
1.3 تیتراسیون پتانسیومتری
تیتراسیون پتانسیومتری در مواردی انجام می شود که شاخص های شیمیایی قابل استفاده نباشد یا شاخص مناسب در دسترس نباشد.
در تیتراسیون پتانسیومتری، از الکترودهای پتانسیومتر قرار داده شده در محلول تیتر شده به عنوان نشانگر استفاده می شود. در این مورد از الکترودهایی استفاده می شود که به یون های تیتر شده حساس هستند. در طی فرآیند تیتراسیون، غلظت یون تغییر می کند که در مقیاس اندازه گیری پتانسیومتر ثبت می شود. پس از ثبت قرائت های پتانسیومتر در واحدهای pH یا mV، وابستگی آنها به حجم تیتراسیون (منحنی تیتراسیون) را ترسیم کنید، نقطه هم ارزی و حجم تیتر مصرفی را برای تیتراسیون تعیین کنید. بر اساس داده های به دست آمده، منحنی تیتراسیون پتانسیومتری ساخته شده است.
منحنی تیتراسیون پتانسیومتری شکلی شبیه به منحنی تیتراسیون در آنالیز تیتریومتری دارد. منحنی تیتراسیون برای تعیین نقطه هم ارزی که در وسط پرش تیتراسیون قرار دارد استفاده می شود. برای این کار، مماس ها به بخش هایی از منحنی تیتراسیون رسم می شوند و نقطه هم ارزی در وسط مماس پرش تیتراسیون تعیین می شود. بزرگترین مقدار تغییر ? pH/?Vدر نقطه هم ارزی به دست می آورد.
نقطه هم ارزی را می توان با دقت بیشتری با روش Gran تعیین کرد که برای ساخت وابستگی استفاده می شود. ? V/?Eاز حجم تیتراتور. با استفاده از روش Gran می توان تیتراسیون پتانسیومتری را بدون رساندن آن به نقطه هم ارزی انجام داد.
تیتراسیون پتانسیومتری در تمام موارد آنالیز تیتریمتری استفاده می شود.
تیتراسیون اسید-باز از یک الکترود شیشه ای و یک الکترود مرجع استفاده می کند. از آنجایی که الکترود شیشه ای به تغییرات pH محیط حساس است، هنگام تیتر شدن آنها، تغییرات pH محیط روی پتانسیومتر ثبت می شود. تیتراسیون پتانسیومتری اسید-باز با موفقیت در تیتراسیون اسیدها و بازهای ضعیف (pK?8) استفاده می شود. هنگام تیتراسیون مخلوط اسیدها، لازم است که pK آنها بیش از 4 واحد متفاوت باشد، در غیر این صورت بخشی از اسید ضعیف تر همراه با اسید قوی تیتر می شود و جهش تیتراسیون به وضوح بیان نمی شود.
این امکان استفاده از پتانسیومتری را برای ساخت منحنیهای تیتراسیون تجربی، انتخاب شاخصهای تیتراسیون و تعیین ثابت اسیدیته و بازی میدهد.
در تیتراسیون پتانسیومتری بارش، از یک الکترود ساخته شده از فلز که با یون های تعیین شده یک جفت الکترود تشکیل می دهد به عنوان نشانگر استفاده می شود.
هنگامی که از تیتراسیون کمپلکس سنجی استفاده می شود: الف) یک الکترود فلزی قابل برگشت به یون فلز در حال تعیین. ب) یک الکترود پلاتین در حضور یک زوج ردوکس در محلول. هنگامی که یکی از اجزای جفت ردوکس توسط تیترانت محدود می شود، غلظت آن تغییر می کند که باعث تغییر در پتانسیل الکترود پلاتین نشانگر می شود. از تیتراسیون مجدد محلول EDTA اضافی اضافه شده به نمک فلزی با محلول نمک آهن (III) نیز استفاده می شود.
برای تیتراسیون ردوکس، یک الکترود مرجع و یک الکترود نشانگر پلاتین، حساس به زوج های ردوکس، استفاده می شود.
تیتراسیون پتانسیومتری به دلیل سادگی، دسترسی، گزینش پذیری و قابلیت های گسترده، یکی از پرکاربردترین روش های آنالیز ابزاری است.
1.4 هدایت سنجی. تیتراسیون هدایت سنجی
هدایت سنجی بر اساس اندازه گیری هدایت الکتریکی یک محلول است. اگر دو الکترود در محلول یک ماده قرار داده شود و اختلاف پتانسیل به الکترودها اعمال شود، جریان الکتریکی از محلول عبور می کند. مانند هر رسانای برق، محلول ها با مقاومت مشخص می شوند آرو کمیت معکوس آن - هدایت الکتریکی L:
جایی که آر- مقاومت، اهم؛
مقاومت ویژه، اهم. سانتی متر؛
اس - مساحت سطح، سانتی متر 2.
جایی که L - هدایت الکتریکی، اهم-1؛
آر- مقاومت، اهم.
تجزیه و تحلیل هدایت سنجی با استفاده از رسانایی سنج ها - دستگاه هایی که مقاومت محلول ها را اندازه گیری می کنند، انجام می شود. با مقدار مقاومت آرتعیین رسانایی الکتریکی محلول ها که مقدار معکوس آن است L.
غلظت محلول ها با هدایت سنجی مستقیم و تیتراسیون هدایت سنجی تعیین می شود. هدایت سنجی مستقیمبرای تعیین غلظت محلول با استفاده از نمودار کالیبراسیون استفاده می شود. برای ایجاد یک نمودار کالیبراسیون، رسانایی الکتریکی یک سری محلول با غلظت مشخص اندازهگیری میشود و نمودار کالیبراسیون هدایت الکتریکی در مقابل غلظت ساخته میشود. سپس رسانایی الکتریکی محلول مورد تجزیه و تحلیل اندازه گیری شده و غلظت آن از نمودار مشخص می شود.
بیشتر استفاده می شود تیتراسیون هدایت سنجی. در این حالت محلول آنالیز شده در یک سلول دارای الکترود قرار می گیرد، سلول روی یک همزن مغناطیسی قرار می گیرد و با تیتر مناسب تیتر می شود. تیترانت در قسمت های مساوی اضافه می شود. پس از افزودن هر بخش از تیترانت، رسانایی الکتریکی محلول را اندازه گیری کنید و رابطه بین رسانایی الکتریکی و حجم تیترانت را رسم کنید. هنگامی که یک تیترانت اضافه می شود، تغییری در هدایت الکتریکی محلول رخ می دهد. منحنی تیتراسیون خم می شود.
از n تحرک یون ها به جریان الکتریکی بستگی دارد رسانایی محلول: تحرک بیشتر است یون های b، الکتریکی بیشتر است رسانایی محلول
تیتراسیون هدایت سنجی چندین مزیت دارد. می توان آن را در محیط های کدر و رنگی و در غیاب شاخص های شیمیایی انجام داد. این روش حساسیت را افزایش داده است و به شما امکان می دهد محلول های رقیق مواد را تجزیه و تحلیل کنید (تا 10-4 mol/dmі). مخلوط مواد با تیتراسیون هدایت سنجی تجزیه و تحلیل می شوند، زیرا تفاوت در تحرک یون های مختلف قابل توجه است و می تواند در حضور یکدیگر به طور متفاوت تیتر شود.
آنالیز هدایت سنجی می تواند به راحتی خودکار شود اگر محلول تیتراتور از یک بورت با سرعت ثابت تامین شود و تغییر در هدایت الکتریکی محلول روی یک ضبط کننده ثبت شود.
این نوع هدایت سنجی نامیده می شود chrono- تجزیه و تحلیل هدایت سنجی.
در تیتراسیون اسید-باز، اسیدهای قوی، اسیدهای ضعیف، نمک بازهای ضعیف و اسیدهای قوی را می توان با هدایت سنجی تعیین کرد.
که در بارشی هدایت سنجیتیتراسیونهدایت الکتریکی محلول های تیتر شده ابتدا به دلیل اتصال الکترولیت تیتر شده به یک رسوب کاهش می یابد یا در یک سطح ثابت باقی می ماند. هنگامی که مقدار بیش از حد تیتر ظاهر می شود، دوباره افزایش می یابد.
که در مجتمعتیتراسیون رسانایی متریکتغییرات در هدایت الکتریکی محلول به دلیل اتصال کاتیون های فلزی به یک کمپلکس با EDTA رخ می دهد.
هدایت سنجی ردوکستیترو- یونبر اساس تغییر در غلظت یون های واکنش دهنده و ظهور یون های جدید در محلول که هدایت الکتریکی محلول را تغییر می دهد.
در سال های اخیر توسعه وجود داشته است هدایت سنجی فرکانس بالا، که در آن الکترودها با محلول تماس نمی گیرند، که هنگام تجزیه و تحلیل رسانه ها و محلول های تهاجمی در رگ های بسته مهم است.
دو گزینه توسعه یافته است - سر راستهدایت سنجی فرکانس بالا و تیتراسیون با فرکانس بالا.
هدایت سنجی با فرکانس بالا مستقیم برای تعیین میزان رطوبت مواد، دانه، چوب، غلظت محلول ها در ظروف بسته - آمپول ها و هنگام تجزیه و تحلیل مایعات تهاجمی استفاده می شود.
تیتراسیون با فرکانس بالا بر روی تیترهای ویژه - TV-6، TV-6L انجام می شود.
تیتراسیون های هدایت سنجی فرکانس بالا به عنوان تیتراسیون اسید-باز، اکسیداسیون و کاهش یا رسوب در مواردی که شاخص مناسب در دسترس نیست یا هنگام تجزیه و تحلیل مخلوط مواد انجام می شود.
1.5 کولومتری. تیتراسیون کولومتریک
در کولومتری، مواد با اندازه گیری مقدار الکتریسیته صرف شده برای تبدیل کمی الکتروشیمیایی آنها تعیین می شوند. تجزیه و تحلیل کولومتریک در یک سلول الکترولیتی انجام می شود که محلولی از ماده تعیین شده در آن قرار می گیرد. هنگامی که یک پتانسیل مناسب به الکترودهای سلول اعمال می شود، کاهش الکتروشیمیایی یا اکسیداسیون ماده رخ می دهد. طبق قوانین الکترولیز که توسط فارادی کشف شد، مقدار ماده ای که در الکترود واکنش نشان می دهد متناسب با مقدار برق عبوری از محلول است:
جایی که g- جرم ماده آزاد شده، گرم؛
n- تعداد الکترون های منتقل شده در فرآیند الکترود؛
اف- عدد فارادی (F = 96485 C/mol)؛
من- قدرت جریان، A؛
t-بار؛
م- جرم مولی ماده آزاد شده، g/mol.
تجزیه و تحلیل کولومتریک امکان تعیین موادی را فراهم می کند که در طی یک واکنش الکتروشیمیایی روی الکترودها رسوب نمی کنند یا به اتمسفر فرار نمی کنند.
کولومتری مستقیم و تیتراسیون کولومتریک وجود دارد.. دقت و حساسیت بالای روشهای اندازهگیری جریان الکتریکی، آنالیز کولومتری را با دقت منحصر به فرد 0.1-0.001% و حساسیت تا 1 10 -8 را ارائه میدهد؟ 1 10 -10 گرم بنابراین، تجزیه و تحلیل کولومتریک برای تعیین ریز ناخالصی ها و محصولات تخریب مواد استفاده می شود، که هنگام نظارت بر کیفیت آنها مهم است.
برای نشان دادن i.e. هنگامی که تیتراسیون کولومتری می توان از روش های شیمیایی و ابزاری استفاده کرد - اضافه کردن شاخص ها، تشخیص ترکیبات رنگی به روش فتومتریک یا اسپکتروفتومتری.
برخلاف سایر روشهای تحلیل، کولومتری میتواند کاملاً خودکار باشد که خطاهای تعیین تصادفی را به حداقل میرساند. این ویژگی برای ایجاد تیتراتورهای کولومتریک خودکار - ابزارهای حساسی که برای تجزیه و تحلیل های دقیق استفاده می شود، زمانی که روش های دیگر به اندازه کافی حساس نیستند، استفاده شد. هنگام تجزیه و تحلیل موادی که در آب محلول اندکی هستند، کولومتری را می توان بر روی الکترودهای سیاه استیلن انجام داد، که جاذب خوبی هستند و چنین موادی را با کاملیت کافی از محیط واکنش حذف می کنند. تیتراسیون کولومتریک روشی امیدوارکننده برای تحلیل ابزاری است. این می تواند کاربرد گسترده ای برای حل تعدادی از مشکلات تحلیلی خاص پیدا کند - تجزیه و تحلیل ناخالصی ها، مقادیر کم دارو، تعیین مواد سمی، عناصر کمیاب و سایر ترکیبات در مواد بیولوژیکی و محیط.
نتیجه
این کار مروری بر روشهای اصلی تحقیق الکتروشیمیایی را ارائه میکند و اصول، کاربرد، مزایا و معایب آنها را به تفصیل شرح میدهد.
روش های الکتروشیمیایی آنالیز گروهی از روش های آنالیز شیمیایی کمی بر اساس استفاده از الکترولیز هستند.
انواع روش عبارتند از آنالیز الکتروگراویمتری (الکتروآنالیز)، الکترولیز داخلی، تبادل تماس فلزات (سیمان)، آنالیز پلاروگرافیک، کولومتری و غیره. به طور خاص، آنالیز الکتروگراویمتری بر اساس وزن کردن ماده آزاد شده روی یکی از الکترودها است. این روش نه تنها امکان تعیین کمی مس، نیکل، سرب و غیره را نیز فراهم می کند، بلکه مخلوطی از مواد را نیز جدا می کند.
علاوه بر این، روش های الکتروشیمیایی آنالیز شامل روش های مبتنی بر اندازه گیری هدایت الکتریکی (رسانایی سنجی) یا پتانسیل الکترود (پتانسیومتری) است. برخی از روش های الکتروشیمیایی برای یافتن نقطه پایانی تیتراسیون (تیتراسیون آمپرومتریک، تیتراسیون هدایت سنجی، تیتراسیون پتانسیومتری، تیتراسیون کولومتری) استفاده می شود.
فهرست منابع استفاده شده
1. مبانی آنالیز الکتروشیمیایی مدرن. بودنیکوف G.K.، Maistrenko V.N.، Vyaselev M.R.، M.، Mir، 2003.
2. جی پلمبک، ویرایش. S. G. Mayranovsky روش های الکتروشیمیایی تجزیه و تحلیل. مبانی نظریه و کاربرد: ترجمه. از انگلیسی / ویدانیا: میر، 1985.
3. Damaskin B.B.، Petriy O.A.، Tsirlina G.A. Electrochemistry - M.: chemistry, 2001. 624 p.
4. STO 005-2015. سیستم مدیریت کیفیت. فعالیت های آموزشی و روش شناختی. ثبت پروژه (آثار) دوره و آثار واجد شرایط نهایی تخصص های فنی.
ارسال شده در Allbest.ru
...اسناد مشابه
طبقه بندی روش های الکتروشیمیایی تجزیه و تحلیل، ماهیت ولتامتری، هدایت سنجی، پتانسیومتری، آمپرومتری، کولومتری، کاربرد آنها در حفاظت از محیط زیست. مشخصات تجهیزات تجزیه شیمیایی و شرکت های فروشنده اصلی.
کار دوره، اضافه شده 01/08/2010
روش های الکتروشیمیایی بر اساس اندازه گیری پارامترهای الکتریکی پدیده های الکتروشیمیایی رخ داده در محلول مورد مطالعه است. طبقه بندی روش های الکتروشیمیایی تجزیه و تحلیل. تیتراسیون پتانسیومتری، هدایت سنجی، کولومتریک.
چکیده، اضافه شده در 01/07/2011
طبقه بندی روش های الکتروشیمیایی تجزیه و تحلیل. تعیین پتانسیومتری غلظت یک ماده در محلول. اصل هدایت سنجی. انواع واکنش ها در طی تیتراسیون هدایت سنجی تجزیه و تحلیل کمی پلاروگرافی. کولومتری مستقیم
کار دوره، اضافه شده در 04/04/2013
ماهیت روش های الکتروتحلیلی، توانایی به دست آوردن اطلاعات تجربی در مورد سینتیک و ترمودینامیک سیستم های شیمیایی است. مزایا، معایب و مناسب بودن ولتامتری، هدایت سنجی، پتانسیومتری، آمپرومتری و کولومتری.
چکیده، اضافه شده در 2009/11/20
مشخصات کلی آنالیز پتانسیومتری الکترودهای نشانگر (تبدیل الکترون و انتخاب یون). انواع روش پتانسیومتری آنالیز. پتانسیومتری مستقیم و تیتراسیون پتانسیومتری. اندازه گیری EMF مدارهای الکتروشیمیایی
کار دوره، اضافه شده 06/08/2012
مفاهیم کلی، شرایط و طبقه بندی روش های الکتروشیمیایی آنالیز. آنالیز پتانسیومتری (پتانسیومتری). تیتراسیون آمپرومتریک (تیتراسیون قطبش پتانسیومتری). تجزیه و تحلیل کمی پلاروگرافی.
چکیده، اضافه شده در 10/01/2012
روش های تحقیق الکتروشیمیایی، طبقه بندی و ماهیت آنها، تاریخچه وقوع. تعیین غلظت اسید با تیتراسیون هدایت سنجی. پتانسیل الکترودها، EMF یک سلول گالوانیکی، معادل الکتروشیمیایی مس.
کار دوره، اضافه شده در 12/15/2014
بررسی روش آنالیز پتانسیومتری. تجزیه و تحلیل و ارزیابی موضوعات تحقیق. مطالعه تکنیک آنالیز پتانسیومتری که در این شیء اعمال می شود. تعیین امکان استفاده از روش های آنالیز پتانسیومتری فرآورده های گوشتی.
کار دوره، اضافه شده در 1396/09/16
روشهای پایه الکتروشیمیایی تجزیه و تحلیل مشخصات کلی آنالیز پتانسیومتری انواع روش پتانسیومتری آنالیز. استفاده از یک سلول گالوانیکی متشکل از دو الکترود. روش اندازه گیری پتانسیل الکترود نشانگر.
کار دوره، اضافه شده در 08/11/2014
طبقه بندی روش های تحلیل ابزاری بر اساس پارامتر در حال تعیین و روش اندازه گیری. جوهر تیتراسیون پتانسیومتری، آمپرومتریک، کروماتوگرافی و فتومتریک. تعیین کمی و کیفی کلرید روی.
روشهای الکتروشیمیایی آنالیز مجموعهای از روشهای آنالیز کمی و کیفی بر اساس پدیدههای الکتروشیمیایی هستند که در محیط مورد مطالعه یا در سطح مشترک رخ میدهند و با تغییرات در ساختار، ترکیب شیمیایی یا غلظت آنالیت مرتبط هستند.
روش های الکتروشیمیایی تجزیه و تحلیل (ECMA) بر اساس فرآیندهایی است که روی الکترودها یا فضای بین الکترودها اتفاق می افتد. مزیت آنها دقت بالا و سادگی نسبی هر دو روش تجهیزات و تجزیه و تحلیل است. دقت بالا توسط الگوهای بسیار دقیق مورد استفاده در ECMA تعیین می شود. یک راحتی بزرگ این است که در این روش از تأثیرات الکتریکی استفاده می شود و این واقعیت که نتیجه این تأثیر (پاسخ) نیز به صورت یک سیگنال الکتریکی به دست می آید. این امر سرعت و دقت بالای خواندن را تضمین می کند و امکانات گسترده ای را برای اتوماسیون باز می کند. ECMA با حساسیت و گزینش پذیری خوب متمایز می شود؛ در برخی موارد می توان آنها را به عنوان میکروآنالیز طبقه بندی کرد، زیرا گاهی اوقات کمتر از 1 میلی لیتر محلول برای تجزیه و تحلیل کافی است.
با توجه به انواع سیگنال های تحلیلی، آنها به موارد زیر تقسیم می شوند:
1) هدایت سنجی - اندازه گیری هدایت الکتریکی محلول آزمایش.
2) پتانسیومتری - اندازه گیری پتانسیل تعادل بدون جریان الکترود نشانگر که ماده آزمایشی در حال تعیین پتانسیل است.
3) کولومتری - اندازه گیری مقدار برق مورد نیاز برای تبدیل کامل (اکسیداسیون یا کاهش) ماده مورد مطالعه.
4) ولتامتری - اندازه گیری ویژگی های قطبش ثابت یا غیر ثابت الکترودها در واکنش های مربوط به ماده آزمایش.
5) وزن سنجی الکتریکی - اندازه گیری جرم ماده آزاد شده از محلول در طول الکترولیز.
27. روش پتانسیومتری.
پتانسیومتری - اندازه گیری پتانسیل تعادل بدون جریان الکترود نشانگر، که ماده آزمایشی برای آن تعیین کننده است.
الف) استاندارد (الکترود مرجع) - دارای پتانسیل ثابت و مستقل از تأثیرات خارجی است. مقررات
ب) الکترود منفرد - پتانسیل آن به غلظت ماده بستگی دارد.
پتانسیل به غلظت بستگی دارد: E = f(c)
معادله نریست E= E° + lna کت
E° - استاندارد الکترون. پتانسیل (پایان)
آر- دانشگاه گاز ثابتپایان)
T – دمای مطلق (تی)- +273 °
.п - تعداد الکترون های درگیر. در اکسیداسیون/کاهش واکنش ها
. الف – تمرکز فعال
روش پتانسیومتری
پتانسیومتری یونومتری (محلول کوچکی به محلول تحقیقاتی اضافه می شود. محلول استاندارد (تیتران) در قسمت هایی اضافه می شود، پس از هر افزودن پتانسیل اندازه گیری می شود. - E)
نقطه هم ارزی
EСх Vх = ل t *Vt
28. روش هدایت سنجی.
هدایت سنجی - اندازه گیری هدایت الکتریکی محلول آزمایش.
تیتراسیون هدایت سنجی
هادی سنج (دستگاه)
آنالیز هدایت سنجی (رسانایی سنجی) بر اساس استفاده از رابطه بین هدایت الکتریکی (رسانایی الکتریکی) محلول های الکترولیت و غلظت آنها است.
رسانایی الکتریکی محلول های الکترولیت - هادی های نوع دوم - بر اساس اندازه گیری مقاومت الکتریکی آنها در یک سلول الکتروشیمیایی که یک ظرف شیشه ای (شیشه ای) با دو الکترود لحیم شده در آن است که محلول الکترولیت آزمایشی بین آنها قرار می گیرد، قضاوت می شود. واقع شده. یک جریان الکتریکی متناوب از سلول عبور می کند. الکترودها اغلب از فلز پلاتین ساخته می شوند که برای افزایش سطح الکترودها، با رسوب الکتروشیمیایی ترکیبات پلاتین از محلول ها (الکترودهای پلاتین شده پلاتین) با لایه ای از پلاتین اسفنجی پوشانده می شود.
29. پلاروگرافی.
پلاروگرافی روشی برای تجزیه و تحلیل شیمیایی کمی و کیفی است که بر اساس به دست آوردن منحنی های جریان در مقابل ولتاژ در مداری متشکل از محلول مورد مطالعه و الکترودهای غوطه ور در آن است که یکی از آنها بسیار قطبش پذیر و دیگری عملاً غیرقابل قطبش است. چنین منحنی ها - پلاروگرام ها - با استفاده از پلاروگراف ها به دست می آیند.
روش پلاروگرافی با حساسیت بالا مشخص می شود. برای انجام آنالیز معمولاً 3-5 میلی لیتر از محلول آزمایش کافی است. تجزیه و تحلیل با استفاده از یک پلاروگراف ضبط خودکار تنها حدود 10 دقیقه طول می کشد. پلاروگرافی برای تعیین محتوای مواد سمی در اشیاء با منشاء بیولوژیکی (به عنوان مثال، ترکیبات جیوه، سرب، تالیم و غیره)، برای تعیین درجه اشباع اکسیژن خون، مطالعه ترکیب هوای بازدمی، استفاده می شود. و مواد مضر موجود در هوای شرکت های صنعتی روش تجزیه و تحلیل پلاروگرافی بسیار حساس است و امکان تعیین مواد در غلظت های بسیار کم (تا 0001/0 درصد) را در محلول فراهم می کند.
30. طبقه بندی روش های تحلیل طیفی. مفهوم طیف.
آنالیز طیفی مجموعه ای از روش ها برای تعیین کیفیت و کمیت است. ترکیب و همچنین ساختار ماده (بر اساس برهمکنش جسم تحقیق با انواع مختلف تابش.)
همه روشهای طیفسنجی بر اساس برهمکنش اتمها، مولکولها یا یونهایی هستند که ماده مورد تجزیه و تحلیل را با تابش الکترومغناطیسی تشکیل میدهند. این برهمکنش خود را در جذب یا گسیل فوتون ها (کوانتا) نشان می دهد. بسته به ماهیت برهمکنش نمونه با تابش الکترومغناطیسی، دو گروه از روش ها متمایز می شوند:
انتشار و جذب. بسته به اینکه کدام ذرات سیگنال تحلیلی را تشکیل می دهند، بین روش های طیف سنجی اتمی و روش های طیف سنجی مولکولی تمایز قائل می شود.
انتشار
در روش های انتشار، نمونه مورد تجزیه و تحلیل در نتیجه تحریک خود فوتون ساطع می کند.
جذب
در روش های جذب، تابش از یک منبع خارجی از نمونه عبور می کند و برخی از کوانتوم ها به طور انتخابی توسط اتم ها یا مولکول ها جذب می شوند.
دامنه- توزیع مقادیر یک کمیت فیزیکی (معمولا انرژی، فرکانس یا جرم). به نمایش گرافیکی چنین توزیعی، نمودار طیفی می گویند. به طور معمول، طیف به طیف الکترومغناطیسی - طیف فرکانس ها (یا همان انرژی های کوانتومی) تابش الکترومغناطیسی اشاره دارد.
1-انعکاس نور
2. چرخش پرتو نور (انحراف)
3. پراکندگی نور: نفرومتری، کدورت سنجی
4-جذب نور
5 انتشار مجدد
الف) فسفرسانس (طولانی طول می کشد)
ب) فلورسانس (بسیار کوتاه)
با توجه به ماهیت توزیع مقادیر فیزیکی، طیف ها می توانند گسسته (خط)، پیوسته (جامد) باشند و همچنین ترکیبی از طیف های گسسته و پیوسته را نشان دهند.
نمونههایی از طیفهای خطی شامل طیفهای جرمی و طیفهای انتقال الکترونیکی پیوندی-پیوندی یک اتم است. نمونه هایی از طیف های پیوسته طیف تابش الکترومغناطیسی یک جامد گرم شده و طیف انتقال های الکترونیکی آزاد یک اتم هستند. نمونههایی از طیفهای ترکیبی، طیفهای نشر ستارهها هستند، که در آن خطوط جذب کرومسفری یا اکثر طیفهای صوتی بر روی طیف پیوسته فتوسفر قرار گرفتهاند.
31. فتومتری: اصل روش، کاربرد در تحقیقات پزشکی قانونی.
نورسنجی - یک روش طیفی مبتنی بر جذب تابش الکترومغناطیسی در محدوده مرئی و نزدیک به فرابنفش (روش مبتنی بر جذب نور است)
اتمی مولکولی
طیف سنجی طیف سنجی (در آنالیز الکترونی)
کووت - نور از آن عبور می کند
ل
I (شدت نور خروجی)
I درجه شدت نور فرودی است.
فوتومتری بخشی از اپتیک فیزیکی و فناوری اندازه گیری است که به روش هایی برای مطالعه ویژگی های انرژی تابش نوری در فرآیند گسیل آن، انتشار در رسانه های مختلف و تعامل با اجسام اختصاص دارد. نورسنجی در محدوده مادون قرمز (طول موج - 10 -3 ... 7 10 -7 متر)، مرئی (7 10 -7 ... 4 10 -7 متر) و فرابنفش (4 10 -7 ...) انجام می شود. 10-8 متر) تابش نوری. هنگامی که تابش الکترومغناطیسی محدوده نوری در یک محیط بیولوژیکی منتشر می شود، تعدادی از اثرات اصلی مشاهده می شود: جذب و پراکندگی تابش توسط اتم ها و مولکول های محیط، پراکندگی ناهمگنی های محیط توسط ذرات، دپلاریزاسیون تابش. با ثبت داده ها در مورد برهمکنش تابش نوری با محیط، می توان پارامترهای کمی مرتبط با ویژگی های پزشکی و بیولوژیکی جسم مورد مطالعه را تعیین کرد. برای اندازه گیری کمیت های فتومتریک از ابزارهایی به نام فتومتر استفاده می شود. از نظر فتومتریک، نور تشعشعی است که در مواجهه با چشم انسان قادر به ایجاد احساس روشنایی است. اساس نورسنجی به عنوان یک علم، نظریه میدان نور است که توسط A. Gershun توسعه یافته است.
دو روش کلی برای فتومتری وجود دارد: 1) نورسنجی بصری، که از توانایی چشم انسان برای درک تفاوت در روشنایی با برابر کردن روشنایی دو میدان مقایسه با ابزارهای مکانیکی یا نوری استفاده می کند. 2) نورسنجی فیزیکی، که در آن از گیرنده های مختلف نور از نوع متفاوت برای مقایسه دو منبع نور - فتوسل های خلاء، فتودیودهای نیمه هادی و غیره استفاده می شود.
32. قانون بوگر-لامبر-بیر، استفاده از آن در تحلیل کمی.
یک قانون فیزیکی که تضعیف یک پرتو تک رنگ موازی نور را هنگام انتشار در یک محیط جاذب تعیین می کند.
قانون با فرمول زیر بیان می شود:
![]() ,
,

شدت پرتو ورودی کجاست، ضخامت لایه ماده ای است که نور از آن عبور می کند، شاخص جذب است (با شاخص جذب بدون بعد که مربوط به فرمول است، طول موج کجاست) اشتباه نشود. .
شاخص جذب ویژگی های یک ماده را مشخص می کند و به طول موج λ نور جذب شده بستگی دارد. این وابستگی را طیف جذبی ماده می گویند.
برای محلول های مواد جاذب در حلال های غیر جاذب نور، شاخص جذب را می توان به صورت زیر نوشت:
جایی که ضریب مشخص کننده برهمکنش یک مولکول یک املاح جذب کننده با نور با طول موج λ، غلظت ماده حل شونده، mol/l است.
بیانیه ای که به آن وابسته نیست، قانون بیر نامیده می شود (با قانون بیر اشتباه نشود). این قانون فرض می کند که توانایی یک مولکول برای جذب نور تحت تأثیر سایر مولکول های اطراف همان ماده در محلول قرار نمی گیرد. با این حال، انحرافات متعددی از این قانون به ویژه در کل مشاهده می شود.
اگر شار نوری با شدت I از لایه خاصی از محلول یا گاز با ضخامت I عبور کند، طبق قانون لامبرت-بیر، مقدار نور جذب شده متناسب با شدت / غلظت c ماده جذب کننده خواهد بود. نور و ضخامت لایه) قانون BMB که شدت تابش نور به ماده و ماده ای که از آن عبور کرده است را با غلظت ماده و ضخامت لایه جذب کننده مرتبط می کند. مانند شکست، فقط تضعیف در ماده. که نور را با درصد معینی جذب می کند. یعنی باقیمانده نور خروجی است
33. طیف سنجی IR.
این روش تجزیه و تحلیل مبتنی بر ثبت طیف جذب مادون قرمز یک ماده است. جذب ماده در ناحیه مادون قرمز به دلیل ارتعاشات اتم ها در مولکول ها اتفاق می افتد. ارتعاشات به کشش (زمانی که فاصله بین اتم ها در طول ارتعاش تغییر می کند) و ارتعاشی (زمانی که زوایای بین پیوندها در طول ارتعاش تغییر می کند) تقسیم می شوند. انتقال بین حالتهای ارتعاشی مختلف در مولکولها کوانتیزه میشود، به همین دلیل جذب در ناحیه IR به شکل یک طیف است که در آن هر ارتعاش طول موج خاص خود را دارد. واضح است که طول موج هر ارتعاش به این بستگی دارد که کدام اتم در آن شرکت می کنند و علاوه بر این، بستگی کمی به محیط آنها دارد.
روش طیف سنجی IR یک روش جداکننده نیست، یعنی در هنگام مطالعه هر ماده ای ممکن است معلوم شود آنچه در واقع مورد مطالعه قرار گرفته مخلوطی از چندین ماده بوده است که البته نتایج رمزگشایی طیف را به شدت مخدوش خواهد کرد. خوب، صحبت در مورد شناسایی بدون ابهام یک ماده با استفاده از روش طیفسنجی IR کاملاً صحیح نیست، زیرا این روش به جای کمیت آنها در یک ترکیب و روش ارتباط آنها با یکدیگر، امکان شناسایی گروههای عملکردی خاص را فراهم میکند.
روش طیفسنجی IR برای انجام تحقیقات بر روی مواد پلیمری، الیاف، پوششهای رنگ و داروهای مخدر استفاده میشود (هنگام شناسایی پرکننده، اغلب از کربوهیدراتها، از جمله پلی ساکاریدها استفاده میشود). این روش به ویژه در مطالعه روان کننده ها ضروری است، زیرا امکان تعیین همزمان ماهیت پایه روان کننده و افزودنی های احتمالی (افزودنی) به این پایه را ممکن می سازد.
34. آنالیز فلورسانس اشعه ایکس.
(XRF) یکی از روش های طیف سنجی مدرن برای مطالعه یک ماده به منظور بدست آوردن ترکیب عنصری آن، یعنی آنالیز عنصری آن است. می توان از آن برای تجزیه و تحلیل عناصر مختلف از بریلیم (Be) تا اورانیوم (U) استفاده کرد. روش XRF بر اساس جمع آوری و تجزیه و تحلیل بعدی طیفی است که با قرار دادن مواد مورد مطالعه در معرض تابش اشعه ایکس به دست می آید. هنگامی که تابش می شود، اتم به حالت برانگیخته می رود، که شامل انتقال الکترون ها به سطوح انرژی بالاتر است. اتم برای مدت بسیار کوتاهی در حالت برانگیخته باقی میماند، در حد یک میکروثانیه، و پس از آن به وضعیت آرام (حالت پایه) باز میگردد. در این حالت، الکترونهای لایههای بیرونی یا جای خالی حاصل را پر میکنند و انرژی اضافی به شکل فوتون ساطع میشود یا انرژی از لایههای بیرونی به الکترون دیگری منتقل میشود (الکترون اوگر).
اکولوژی و حفاظت از محیط زیست: تعیین فلزات سنگین در خاک، رسوبات، آب، ذرات معلق در هوا و غیره.
زمین شناسی و کانی شناسی: تجزیه و تحلیل کمی و کیفی خاک، کانی ها، سنگ ها و غیره.
متالورژی و صنایع شیمیایی: کنترل کیفیت مواد اولیه، فرآیند تولید و محصولات نهایی
صنعت رنگ و لاک: تجزیه و تحلیل رنگ های سرب
35. طیف سنجی گسیل اتمی.
آنالیز طیفی گسیل اتمی مجموعه ای از روش های آنالیز عنصری است که بر اساس مطالعه طیف انتشار اتم ها و یون های آزاد در فاز گاز است. به طور معمول، طیف انتشار در راحت ترین محدوده طول موج نوری از 200 تا 1000 نانومتر ثبت می شود.
AES (طیفسنجی انتشار اتمی) روشی برای تعیین ترکیب عنصری یک ماده از طیفهای نشر نوری اتمها و یونهای نمونه مورد تجزیه و تحلیل، برانگیختهشده در منابع نوری است. به عنوان منابع نور برای تجزیه و تحلیل انتشار اتمی، از شعله مشعل یا انواع پلاسما استفاده می شود، از جمله جرقه الکتریکی یا پلاسمای قوس الکتریکی، پلاسمای جرقه لیزری، پلاسمای جفت شده القایی، تخلیه درخشش و غیره. AES رایج ترین روش سریع و بسیار حساس برای شناسایی و تعیین کمیت عناصر ناخالصی در مواد گازی، مایع و جامد، از جمله موارد با خلوص بالا.
زمینه های استفاده:
متالورژی: تجزیه و تحلیل ترکیب فلزات و آلیاژها،
صنعت معدن: مطالعه نمونه های زمین شناسی و مواد خام معدنی،
اکولوژی: تجزیه و تحلیل آب و خاک،
تجهیزات: تجزیه و تحلیل روغن موتور و سایر مایعات فنی برای ناخالصی های فلزی،
تحقیقات بیولوژیکی و پزشکی.
اصول کارکرد، اصول جراحی، اصول عملکرد.
اصل عملکرد یک طیف سنج نشر اتمی بسیار ساده است. بر اساس این واقعیت است که اتم های هر عنصر می توانند نورهایی با طول موج های خاص - خطوط طیفی ساطع کنند و این طول موج ها برای عناصر مختلف متفاوت است. برای اینکه اتمها شروع به انتشار نور کنند، باید با گرما، تخلیه الکتریکی، لیزر یا وسایل دیگر برانگیخته شوند. هر چه تعداد اتم های یک عنصر معین در نمونه مورد تجزیه و تحلیل بیشتر باشد، تابش طول موج مربوطه روشن تر خواهد بود.
شدت خط طیفی عنصر مورد تجزیه و تحلیل، علاوه بر غلظت عنصر مورد تجزیه و تحلیل، به تعداد زیادی از عوامل مختلف بستگی دارد. به همین دلیل، محاسبه نظری رابطه بین شدت خط و غلظت عنصر مربوطه غیرممکن است. به همین دلیل است که نمونه های استانداردی که از نظر ترکیب مشابه با نمونه مورد تجزیه و تحلیل هستند برای آنالیز مورد نیاز است. این نمونه های استاندارد ابتدا بر روی دستگاه در معرض دید (سوختن) قرار می گیرند. بر اساس نتایج حاصل از این سوختگی ها، یک نمودار کالیبراسیون برای هر عنصر آنالیز شده ساخته می شود. وابستگی شدت خط طیفی یک عنصر به غلظت آن. پس از آن، هنگام تجزیه و تحلیل نمونه ها، از این نمودارهای کالیبراسیون برای محاسبه مجدد شدت های اندازه گیری شده در غلظت ها استفاده می شود.
آماده سازی نمونه ها برای آنالیز.
باید در نظر داشت که در واقعیت چندین میلی گرم از یک نمونه از سطح آن تجزیه و تحلیل می شود. بنابراین، برای به دست آوردن نتایج صحیح، نمونه باید از نظر ترکیب و ساختار همگن باشد و ترکیب نمونه باید با ترکیب فلز مورد تجزیه و تحلیل یکسان باشد. هنگام آنالیز فلز در ریخته گری یا ذوب، توصیه می شود از قالب های مخصوص برای ریخته گری نمونه ها استفاده شود. در این حالت، شکل نمونه می تواند دلخواه باشد. فقط لازم است که نمونه مورد تجزیه و تحلیل سطح کافی داشته باشد و بتوان آن را در پایه محکم کرد. برای آنالیز نمونه های کوچک مانند میله ها یا سیم ها می توان از آداپتورهای ویژه استفاده کرد.
مزایای روش:
بدون تماس،
امکان تعیین کمیت همزمان تعداد زیادی عنصر،
دقت بالا،
محدودیت های تشخیص پایین،
سهولت آماده سازی نمونه،
کم هزینه.
36. طیف سنجی جذب اتمی.
روشی برای تعیین کمی ترکیب عنصری یک ماده مورد مطالعه با استفاده از طیف های جذب اتمی، بر اساس توانایی اتم ها برای جذب انتخابی تابش الکترومغناطیسی در تجزیه. بخش هایی از طیف A.-a.a. در ویژه انجام شد دستگاه ها - جذب اسپکتروفتومترها نمونه ای از مواد مورد تجزیه و تحلیل حل می شود (معمولاً با تشکیل نمک). محلول به شکل آئروسل به شعله مشعل وارد می شود. تحت تأثیر شعله (3000 درجه سانتیگراد)، مولکول های نمک به اتم ها تجزیه می شوند که می توانند نور را جذب کنند. سپس یک پرتو نور از شعله مشعل عبور می کند که در طیف آن خطوط طیفی مربوط به یک یا عنصر دیگر وجود دارد. خطوط طیفی مورد مطالعه با استفاده از یک تک رنگ از تابش کل جدا می شوند و شدت آنها توسط یک واحد ضبط ثبت می شود. ریاضی. پردازش طبق فرمول انجام می شود: J = J0 * e-kvI،
که در آن J و J0 شدت نور عبوری و فرودی است. kv – ضریب جذب، بسته به فرکانس آن؛ I - ضخامت لایه جذب کننده
حساس تر از نیروگاه های هسته ای
37. نفرومتری و کدورت سنجی.
S = log (I°/I) شدت سقوط. در محلول (I°) تقسیم بر شدت خروجی محلول (I) =
کدورت k-const
ب – طول مسیر پرتو نور
N تعداد ذرات در واحد است. راه حل
تجزیه و تحلیل نفلومتری و کدورت سنجی از پدیده پراکندگی نور توسط ذرات جامد معلق در محلول استفاده می کند.
نفلومتری روشی برای تعیین پراکندگی و غلظت سیستم های کلوئیدی بر اساس شدت نور پراکنده شده توسط آنها است. نفرومتری، اندازهگیریها در یک دستگاه خاص، یک نفرومتر، انجام میشود که عملکرد آن بر اساس مقایسه شدت نور پراکنده شده توسط محیط مورد مطالعه با شدت نور پراکنده شده توسط محیط دیگری است که به عنوان یک استاندارد عمل میکند. تئوری پراکندگی نور توسط سیستم های کلوئیدی که در آن اندازه ذرات از نیم طول موج نور فرودی تجاوز نمی کند توسط فیزیکدان انگلیسی J. Rayleigh در سال 1871 ارائه شد. طبق قانون ریلی، شدت نوری که من در جهتی عمود بر پراکنده می کنم. پرتو فرودی با فرمول I = QNvlk بیان می شود - که q شدت نور فرودی است، N تعداد کل ذرات در واحد حجم، یا غلظت جزئی، v حجم یک ذره، \ طول موج است. نور فرودی، k بسته به ضریب شکست ذرات کلوئیدی و محیط پراکندگی اطراف آنها، فاصله از منبع نور و همچنین واحدهای اندازه گیری پذیرفته شده ثابت است.
کدورت سنجی روشی برای آنالیز محیط های کدر بر اساس اندازه گیری شدت نور جذب شده توسط آنهاست. اندازه گیری های کدورت سنجی در نور عبوری با استفاده از کدورت سنج های بصری یا رنگ سنج های فوتوالکتریک انجام می شود. روش اندازه گیری شبیه به روش رنگ سنجی است و بر اساس کاربرد قانون بوگر-لامبرت برای محیط های کدر است که در مورد سوسپانسیون ها فقط برای لایه های بسیار نازک یا در رقت های قابل توجه معتبر است. کدورت سنجی مستلزم رعایت دقیق شرایط برای تشکیل فاز پراکنده است، مشابه شرایط مشاهده شده برای نفلومتری. یک پیشرفت قابل توجه در کدورت سنجی استفاده از تیتراسیون پیک کدورت با استفاده از رنگ سنج های فوتوالکتریک است. کدورت سنجی با موفقیت برای تعیین تحلیلی سولفات ها، فسفات ها، کلریدها، سیانیدها، سرب، روی و غیره استفاده می شود.
مزیت اصلی روشهای نفلومتری و کدورتسنجی حساسیت بالای آنها است که بهویژه در رابطه با عناصر یا یونهایی که هیچ واکنش رنگی برای آنها وجود ندارد، ارزشمند است. به عنوان مثال، در عمل، تعیین نفلومتریک کلرید و سولفات در آب های طبیعی و اشیاء مشابه به طور گسترده مورد استفاده قرار می گیرد. از نظر دقت، کدورت سنجی و نفرومتری نسبت به روش های فتومتریک پایین تر هستند که عمدتاً به دلیل دشواری های بدست آوردن سوسپانسیون با اندازه ذرات یکسان، پایداری در طول زمان و غیره است. علاوه بر خطاهای نسبتاً کوچک معمول در تعیین فتومتریک، خطاهای مرتبط با تکرار ناکافی روش های تجزیه شیمیایی، خواص سوسپانسیون ها اضافه می شود.
به عنوان مثال، از nephelometry و کدورت سنجی برای تعیین SO4 به شکل سوسپانسیون BaSO4، Cl- به شکل سوسپانسیون AgCl، S2- به شکل سوسپانسیون CuS از پایین استفاده می شود. محدودیت محتویات قابل تشخیص ~ 0.1 میکروگرم در میلی لیتر است. برای استاندارد کردن شرایط آنالیز در آزمایشها، کنترل دقیق دما، حجم سوسپانسیون، غلظت معرفها، سرعت هم زدن و زمان اندازهگیری ضروری است. بارش باید به سرعت انجام شود و ذرات رسوب داده شده باید اندازه کوچک و pH پایین داشته باشند. برای جلوگیری از انعقاد ذرات بزرگ، به عنوان مثال، یک تثبیت کننده اغلب به محلول اضافه می شود. ژلاتین، گلیسیرین.
38. کروماتوگرافی: تاریخچه پیدایش، اصل روش، کاربرد در دادگاه. پژوهش.
کروماتوگرافی یک روش جذب دینامیکی برای جداسازی و آنالیز مخلوط مواد و همچنین مطالعه خواص فیزیکوشیمیایی مواد است. این بر اساس توزیع مواد بین دو فاز - ثابت (فاز جامد یا مایع محدود شده به یک حامل بی اثر) و متحرک (فاز گاز یا مایع، شوینده) است. نام این روش با اولین آزمایشهای کروماتوگرافی مرتبط است، که طی آن، توسعهدهنده روش، میخائیل تسوت، رنگدانههای گیاهی با رنگ روشن را جدا کرد.
روش کروماتوگرافی اولین بار توسط گیاه شناس روسی میخائیل سمنوویچ تسوت در سال 1900 استفاده شد. او از ستونی پر از کربنات کلسیم برای جداسازی رنگدانه های گیاهی استفاده کرد. اولین گزارش در مورد توسعه روش کروماتوگرافی توسط Tsvet در 30 دسامبر 1901 در یازدهم کنگره طبیعت شناسان و پزشکاندر سن پترزبورگ اولین کار چاپی در مورد کروماتوگرافی در سال 1903 در مجله منتشر شد مجموعه مقالات انجمن طبیعت گرایان ورشو. ترم اول کروماتوگرافیدر سال 1906 در دو اثر چاپی رنگ که در یک مجله آلمانی منتشر شد ظاهر شد Berichte der Deutschen Botanischen Gesellschaft. در سال 1907، تسوت روش خود را نشان داد انجمن گیاه شناسی آلمان.
در سالهای 1910-1930، این روش بهطور غیرمستقیم فراموش شد و عملاً توسعه پیدا نکرد.
در سال 1931، R. Kuhn، A. Winterstein و E. Lederer، با استفاده از کروماتوگرافی، فراکسیون α و β را به شکل کریستالی از کاروتن خام جدا کردند، و بدین وسیله ارزش آماده سازی روش را نشان دادند.
در سال 1941، A. J. P. Martin و R. L. M. Singh نوع جدیدی از کروماتوگرافی را توسعه دادند که بر اساس تفاوت در ضرایب توزیع مواد جدا شده بین دو مایع غیرقابل اختلاط بود. روش نام گرفت " کروماتوگرافی پارتیشن».
در سال 1947، T. B. Gapon، E. N. Gapon و F. M. Shemyakin روش "کروماتوگرافی تبادل یونی" را توسعه دادند.
در سال 1952، جی. مارتین و آر. سینگ جایزه نوبل شیمی را برای ایجاد روش کروماتوگرافی پارتیشن دریافت کردند.
از اواسط قرن بیستم تا به امروز، کروماتوگرافی به شدت توسعه یافته و به یکی از پرکاربردترین روش های تحلیلی تبدیل شده است.
طبقه بندی: گاز، مایع
مبانی کروماتوگرافی روند.برای انجام کروماتوگرافی جداسازی مواد یا تعیین فیزیکی و شیمیایی آنها. معمولاً از خصوصیات ویژه استفاده می شود. دستگاه ها - کروماتوگرافی. پایه ای واحدهای کروماتوگرافی - کروماتوگرافی. ستون، آشکارساز و دستگاه تزریق نمونه. ستون حاوی جاذب وظیفه جداسازی مخلوط تجزیه شده را به اجزای تشکیل دهنده آن و آشکارساز وظیفه جداسازی مقادیر آنها را انجام می دهد. تعاریف آشکارساز واقع در خروجی ستون به طور خودکار غلظت ترکیبات جدا شده را تعیین می کند. در جریان فاز متحرک پس از معرفی مخلوط تجزیه شده با جریان فاز متحرک به ستون، مناطق همه مواد در ابتدای کروماتوگرافی قرار می گیرند. ستون ها (شکل 1). تحت تأثیر جریان فاز متحرک، اجزای مخلوط شروع به حرکت در امتداد ستون با تجزیه می کنند. سرعت هایی که مقادیر آنها با ضرایب توزیع K اجزای کروماتوگرافی شده نسبت معکوس دارد. موادی که به خوبی جذب شده اند، مقادیر ثابت توزیع برای آنها زیاد است، در امتداد لایه جاذب در امتداد ستون کندتر از موادی که جذب ضعیفی دارند حرکت می کنند. بنابراین، جزء A سریعترین ستون را ترک می کند، سپس جزء B، و آخرین موردی که ستون را ترک می کند جزء C است (K A<К Б <К В). Сигнал детектора, величина к-рого пропорциональна концентрации определяемого в-ва в потоке элюента, автоматически непрерывно записывается и регистрируется (напр., на диаграммной ленте). Полученная хроматограмма отражает расположение хроматографич. зон на слое сорбента или в потоке подвижной фазы во времени.

برنج. 1.جداسازی مخلوطی از سه جزء (A، B و C) روی ستون کروماتوگرافی K با آشکارساز D: a - موقعیت مناطق کروماتوگرافی اجزای جدا شده در ستون در فواصل زمانی معین. b - کروماتوگرام (سیگنال C، زمان t) .
با کروماتوگرافی لایه مسطح هنگام جداسازی، یک ورق کاغذ یا یک صفحه با یک لایه جاذب با نمونه های اعمال شده از ماده مورد مطالعه در کروماتوگرافی قرار می گیرد. دوربین. پس از جداسازی، اجزاء با هر روش مناسب تعیین می شوند.
39. طبقه بندی روش های کروماتوگرافی.
کروماتوگرافی روشی برای جداسازی و تجزیه و تحلیل مواد بر اساس توزیع آنالیت است. تفاوت بین 2 فاز: متحرک و ثابت
محلولی از مخلوطی از موادی که قرار است جدا شوند از یک لوله شیشه ای (ستون Adsorption) پر از جاذب عبور داده می شود. در نتیجه، اجزای مخلوط در ارتفاعات مختلف ستون جاذب به صورت مناطق (لایه) مجزا حفظ می شوند. مواد بهتر از adsorbir است. آنها در بالای ستون هستند و بدتر در پایین ستون جذب می شوند. موادی که نمی توانند جذب شوند بدون توقف از ستون عبور می کنند و در فیلتر جمع می شوند.
طبقه بندی ها:
1. با توجه به حالت تجمع فازها.
1) متحرک
الف) گاز (گازهای بی اثر: هلیوم، آرگون، آزون)
ب) مایع
2. با توجه به روش اجرا
1) در یک هواپیما (مسطح)؛ کاغذ لایه نازک
2) ستونی
الف) بسته بندی شده (ستون بسته بندی شده پر از جاذب)
ب) مویرگی (مویرگی شیشه ای نازک/کوارتز که در سطح داخلی آن یک فاز ثابت اعمال می شود)
شما می توانید دفاع کنید. اقلام در مقادیر کم.
مواد فرار جدا می شوند.
40. کروماتوگرام. پارامترهای اساسی پیک کروماتوگرافی.
کروماتوگرام نتیجه ثبت وابستگی غلظت اجزاء در خروجی ستون به زمان است.
اچ اس
هر پیک در کروماتوگرام با دو مشخصه مشخص می شود پارامترهای اصلی
1. زمان نگهداری ( تی آر) زمان از لحظه معرفی نمونه آنالیز شده تا ثبت حداکثر پیک کروماتوگرافی است. این بستگی به ماهیت ماده دارد و یک ویژگی کیفی است.
2. ارتفاع ( ساعت) یا منطقه ( اس) اوج
اس = ½ ω × ساعت. (4)
ارتفاع و مساحت قله به مقدار ماده بستگی دارد و از مشخصات کمی است.
زمان ماند شامل دو جزء است - زمان ماندن مواد در فاز متحرک ( تی متر) و زمان اقامت در فاز ثابت ( تی س):
شناسایی قله های اجزای ناشناخته مخلوط تجزیه شده با مقایسه (مقایسه) انجام می شود. مقادیر مستقیماً از کروماتوگرام با داده های جدول بندی شده مربوط به ترکیبات شناخته شده تعیین می شود. هنگام شناسایی در کروماتوگرافی، فقط منفی قابل اعتماد است. پاسخ؛ برای مثال، اگر زمانهای نگهداری پیک i و آیتم A با هم مطابقت نداشته باشند، پیک i مورد A نیست. همزمانی زمان های نگهداری پیک i و مورد الف شرط لازم اما کافی نیست تا نتیجه گیری کنیم که اوج i مورد A است.
در کار عملی، انتخاب یک یا آن پارامتر برای تفسیر کمی کروماتوگرام ها با تأثیر ترکیبی چندین عامل تعیین می شود: سرعت و سهولت محاسبه، شکل (عریض، باریک) و درجه عدم تقارن پیک کروماتوگرافی، کارایی ستون مورد استفاده، کامل بودن جداسازی اجزای مخلوط، وجود دستگاه های خودکار لازم (ادغام کننده ها، سیستم های کامپیوتری برای پردازش داده های کروماتوگرافی).
پارامتر تعیین شده پیک کروماتوگرافی توسط اپراتور بر روی کروماتوگرام در پایان چرخه جداسازی اجزای مخلوط تجزیه شده به صورت دستی اندازه گیری می شود.
پارامتر تعیین شده پیک کروماتوگرافی به طور خودکار با استفاده از ولت مترهای دیجیتال، یکپارچه سازها یا رایانه های تخصصی به طور همزمان با جداسازی اجزای مخلوط تجزیه شده در ستون و ثبت کروماتوگرام اندازه گیری می شود.
از آنجایی که تکنیک رمزگشایی کروماتوگرام ها به اندازه گیری پارامترهای پیک های کروماتوگرافی ترکیب مورد نظر و ترکیب استاندارد خلاصه می شود، شرایط کروماتوگرافی باید جداسازی کامل آنها را تا حد امکان تضمین کند؛ تمام اجزای دیگر نمونه اصلی تحت شرایط آنالیز پذیرفته شده ممکن است از یکدیگر جدا نشوند و یا اصلاً روی کروماتوگرام ظاهر نشوند (این مزیت روش استاندارد داخلی نسبت به روش عادی سازی داخلی است)
41. تجزیه و تحلیل کروماتوگرافی کیفی.
با طول ستون کافی می توان به جداسازی کامل اجزای هر مخلوطی دست یافت. و پس از شستشوی اجزای جدا شده به فراکسیون های جداگانه (Eluates)، تعداد اجزای مخلوط را تعیین کنید (مطابق با تعداد مایعات است)، ترکیب کیفی آنها را تعیین کنید، مقدار هر یک از آنها را با استفاده از روش های مناسب تجزیه و تحلیل کمی تعیین کنید. .
تجزیه و تحلیل کروماتوگرافی کیفی، به عنوان مثال. شناسایی یک ماده با توجه به کروماتوگرام آن را می توان با مقایسه ویژگی های کروماتوگرافی، اغلب حجم باقیمانده (یعنی حجم فاز متحرک عبور از ستون از ابتدای ورودی مخلوط تا ظاهر شدن این جزء در خروجی انجام داد. از ستون)، تحت شرایط خاصی برای اجزای مخلوط مواد تجزیهشده و برای استاندارد یافت میشود.
42. تجزیه و تحلیل کروماتوگراف کمی.
آنالیز کمّی کروماتوگرافی معمولاً بر روی کروماتوگرافی انجام می شود. این روش بر اساس اندازهگیری پارامترهای مختلف پیک کروماتوگرافی بسته به غلظت مواد کروماتوگرافی - ارتفاع، عرض، سطح و حجم باقیمانده یا محصول حجم باقیمانده و ارتفاع پیک است.
در کروماتوگرافی گازی کمی از روش های کالیبراسیون مطلق و نرمال سازی داخلی یا نرمال سازی استفاده می شود. از روش استاندارد داخلی نیز استفاده می شود. با کالیبراسیون مطلق، وابستگی ارتفاع یا مساحت قله به غلظت ماده به صورت تجربی تعیین می شود و نمودارهای کالیبراسیون ساخته می شوند یا ضرایب مربوطه محاسبه می شوند. سپس، همان ویژگیهای پیکها در مخلوط آنالیز شده تعیین میشوند و غلظت آنالیت از نمودار کالیبراسیون پیدا میشود. این روش ساده و دقیق، اصلی ترین روش برای تعیین ناخالصی های کمیاب است.
هنگام استفاده از روش نرمال سازی داخلی، مجموع پارامترهای قله، به عنوان مثال، مجموع ارتفاعات همه قله ها یا مجموع مساحت آنها، 100٪ در نظر گرفته می شود. سپس نسبت ارتفاع یک قله منفرد به مجموع ارتفاعات یا نسبت مساحت یک قله به مجموع مساحت ها وقتی در 100 ضرب شود، کسر جرمی (%) جزء را مشخص می کند. مخلوط با این رویکرد، لازم است که وابستگی مقدار پارامتر اندازه گیری شده به غلظت برای همه اجزای مخلوط یکسان باشد.
43. کروماتوگرافی مسطح. استفاده از کروماتوگرافی لایه نازک برای آنالیز جوهر.
اولین شکل استفاده از سلولز در کروماتوگرافی لایه نازک، کروماتوگرافی کاغذی بود. صفحات TLC موجود و TLC با کارایی بالا امکان جداسازی مخلوطهای مواد قطبی را با استفاده از حداقل مخلوطهای سه تایی آب، یک حلال آلی غیرقابل اختلاط و یک حلال محلول در آب که تشکیل یک فاز را به عنوان مواد شوینده تقویت میکند، امکان پذیر میسازد.