Elektrokémiai módszerek– a legdinamikusabban fejlődő környezetmonitoring alkalmazásukat tekintve. A MOS rendszerekben leggyakrabban használt módszerek a voltammetria (beleértve a polarográfiát), a potenciometria (beleértve az ionometriát), a coulometria és a konduktometria.
Az elektrokémiai elemzési módszerek a közeg különféle elektromos tulajdonságainak a benne vizsgált anyagok mennyiségi tartalmától és minőségi összetételétől való függését használják fel:
· változás lehetséges elektródát az anyagban végbemenő fizikai és kémiai folyamatoktól függően ( potenciometrikus módszer), beleértve a ion-szelektív elektródák szelektív reakciói, amelyek egyedileg érzékenyek nagyszámú kationra és anionra ( ionometrikus módszer);
· változás elektromos vezetőképesség (áram)és egy anyag dielektromos állandója a közeg természetétől és összetevőinek koncentrációjától függően konduktometrikusÉs amperometrikus mód);
· változtatások villamos energia mennyisége amikor az analit bejut az elektrokémiai cellába ( coulometriás módszer);
· a vizsgált vegyület visszanyerése higanycsepegő vagy forgó elektródán, általában, ha nyomokban különböző aggregációs állapotú anyagokat vizsgálunk ( polarográfiai vagy voltammetrikus módszer).
Az ebbe a csoportba tartozó összes eszköz polarográfja a legmagasabb érzékenységgel rendelkezik, ami 0,005–1 μg/ml minta.
Voltammetria polarizációs görbék vizsgálatán alapuló elektrokémiai elemzési módszerek egy csoportját tartalmazza. Ezek a módszerek polarográfiaÉs amperometrikus titrálás – számos változata és módosítása van. Leggyakoribb állandó áram polarográfia.
A polarográfiai berendezés egyenáramforrásból, feszültségosztóból, ejtő (általában higany) vagy forgó elektródából és egy segédelektródából (általában higany vagy egyéb) áll. Az áramerősség méréséhez egy mikroampermérőt csatlakoztatunk a rendszerhez. Az elektródákat a vizsgálati oldattal együtt elektrolizátorba (cellába) helyezzük.
Az elektrolitikus cellára adott feszültség az anód és a katód polarizációját okozza E= f a– f k +iR, Ahol én– áramerősség; NAK NEK - oldatállóság; f aés f k– az anód és a katód potenciálja.
Ha csökkenti az oldat ellenállását erős elektrolit hozzáadásával (háttér), akkor az érték iR(potenciális oldatcsökkenés) elhanyagolható.
Az anódpotenciál gyakorlatilag állandó marad a cella működése során, mivel az áramsűrűség alacsony, és az anód viszonylag nagy felülete nem polarizált. Ekkor egy kis felületű csöpögő polarizáló katód potenciálja egyenlő lesz: E= -f k. A polarográfiás méréseknél gyakran az edény alján lévő higanyréteg helyett egy nem polarizáló telített kalomelelektródát használnak, amelynek potenciálját nullával egyenlőnek veszik.
A polarográfiai adatokat az elektrolitikus cellán áthaladó áram mérésével nyerjük az elektródákra adott potenciál függvényében. Az áram potenciáltól való grafikus függését polarográfiai hullámnak nevezzük. rizs. 2).
Az elektrolízis kezdetén az EMF alacsony értékeinél az áramerősség szinte állandó lesz, és csak nagyon lassan növekszik. Ez az úgynevezett maradék áram, amely az elektrolízis során végig megmarad.
Rizs. 2. 10–3 M cink-klorid oldat és 1 M kálium-klorid oldat (1. görbe) és 1 M kálium-klorid oldat (2. görbe) polarogramja
Amint az ionredukciós potenciált elérjük (például a meghatározott cinkionoknál ez egyenlő -1,0 V), kisülésük egy higanycseppen kezdődik:
Zn 2+ + 2 +Hg ® Zn (Hg).
A katódon híg cink-amalgám Zn (Hg) képződik, amely amint a lehulló csepp érintkezésbe kerül az anóddal, összetevőire bomlik:
Zn (Hg) – 2 ® Zn 2+ +Hg.
A cinkionok redukciós potenciáljánál az áramerősség meredeken növekszik ( rizs. 2), de egy bizonyos érték elérése után az alkalmazott EMF növekedése ellenére szinte állandó marad. Ezt az áramot korlátozónak vagy diffúziónak nevezik, értéke általában arányos a meghatározandó anyag koncentrációjával.
A polarogramok készítésekor a vizsgált elektrolithoz közömbös elektrolitot adnak, amelynek kationjai sokkal nehezebben redukálódnak, mint az elemzett kation, például KCl, KNO 3, NH 4 Cl; 100-1000-szer nagyobb koncentrációban, mint a meghatározandó anyag koncentrációja. Ezt az elektrolitot „háttérnek” nevezik. A tesztoldatban az elektromos vezetőképesség növelésére és az indikátorelektróda (katód) elektromos mezőjének árnyékolására jön létre. Ezért az analit kationjait nem vonzza a katód elektromos mezeje, hanem diffúzió következtében elmozdulnak felé.
A polarogram legfontosabb jellemzője a félhullámpotenciál E 1/2 és polarográfiai hullámmagasság h(határ diffúziós áram). A félhullám potenciált használják fel minőség polarográfiai elemzés. Különböző anyagok félhullámpotenciáljai, növekvő negatív érték szerint rendezve, alkotják az úgynevezett „polarográfiai spektrumot”. Mivel a félhullámpotenciál jelentősen függ az oldat (a vizsgálandó közeg) összetételétől, a hátteret mindig polarográfiai táblázatokban tüntetjük fel.
BAN BEN mennyiségi A polarográfiai elemzés során a kalibrációs grafikon módszereit, az adalékokat, az összehasonlításokat és a számítási módszereket alkalmazzák a koncentráció mérésére.
A polarográfia különféle lehetőségei közül a módszer differenciális impulzus polarográfia (DIP) ) a leghatékonyabb a környezeti monitoring problémák megoldására, elsősorban nagy érzékenysége miatt. A DIP módszer lehetővé teszi, hogy értékelje a klasszikus polarográfiával meghatározott összes anyag tartalmát. Egyéb polarográfiás módszerek mellett különösen alkalmas nyomelemzésre négyzethullám polarográfia, amely a DIP-hez közeli kimutatási határt biztosít, de csak reverzibilis elektródos eljárások esetén, ezért ezt a módszert gyakran alkalmazzák nehézfémnyomok meghatározására. A DIP módszerrel az elektróda kettős elektromos rétegének kapacitását megváltoztató felületaktív anyagok is meghatározhatók.
Módszerekkel meghatározható a nehézfém-ionok mikrotartalma inverziós elektrokémiai elemzés (IEA) vagy más módon, sztrippelő voltammetriás analízis (IVA ), amelyben a meghatározandó fémek előre lerakódnak az elektródára, majd a polarográfiás ellenőrzés során feloldódnak. Ez az opció a DIP-vel kombinálva az egyik legérzékenyebb elektrokémiai elemzési módszer. Az IEA (IVA) hardver kialakítása viszonylag egyszerű, ami lehetővé teszi a terepen történő elemzések elvégzését, ezen az elven működhetnek az automatizált folyamatos vezérlő (monitoring) állomások is.
Az IEA (IVA) módszerekkel Cu, Pb, Bi, Sb, As, Sn In, Ga, Ag, Tl, Cd, Zn, Hg, Au, Ge, Te, Ni, Co ionok és sok anion meghatározható. Az IEA (IEA) módszerek egyik fontos előnye (ellentétben más módszerekkel, például az atomabszorpciós spektrometriával) képes megkülönböztetni a szabad ionokat a kötött kémiai formáiktól, ami a vizsgált anyagok fizikai-kémiai tulajdonságainak környezetanalitikai ellenőrzés szempontjából (például a vízminőség felmérésekor) történő értékeléséhez is fontos. Számos szerves anyag IEA (IEA) módszerekkel is meghatározható az elektróda felületén való adszorpciós felhalmozódásuk után.
Polarográfiai módszerekkel az ipari helyiségek atmoszférájában és levegőjében lévő különböző fémek aeroszoljai is meghatározhatók, miután azokat megfelelő szűrőkön felfogják, majd a koncentrátumokat oldatba juttatják. A légkörben gázok és gőzök formájában jelenlévő szerves vegyületek polarográfiás módszerrel határozhatók meg, miután speciálisan kiválasztott oldatokkal felszívták őket. A biológiai anyagokban található fémeket és különféle vegyületeket extrakciójuk után általában polarográfiás módszerrel határozzák meg. Minden polarográfiás mérés, beleértve az IEA-t (IVA), teljesen automatizálható, ami elengedhetetlen a soros elemzések elvégzéséhez.
A polarográfia egyik legfontosabb alkalmazási területe a víz oxigéntartalmának meghatározása. Erre a célra amperometrikus detektorokat használnak, amelyek az oldat oxigénkoncentrációjával arányos áramot állítanak elő.
A detektor membrán felületére enzimet alkalmazva különböző enzimamperometrikus érzékelőket lehet előállítani, amelyek alkalmasak biokémiai és klinikai elemzésekre. Az ilyen érzékelőket a környezetfelügyeleti rendszerekben is használják.
Az elektrokatalitikus elven működő elektródák alkalmasak különféle gázok (SO 2, H 2 S, CO, NO x) megfigyelésére az ipari helyiségek levegőjében. Ezeknek a gázoknak az elektróda felületén lezajló (katalizátorszerepet játszó) elektrokémiai reakciói az elektródarendszerben olyan áramot hoznak létre, amely funkcionálisan összefügg a levegőben lévő gázok koncentrációjával.
A polarográfia alkalmazása nem korlátozódik a különálló minták elemzésére, a módszer fokozatosan áttér a gázok és folyadékok folyamatos elemzésének elvei felé.
A voltametriás polarográfiai detektorokat sikeresen alkalmazzák a nagy teljesítményű folyadékkromatográfiában (HPLC). Ebben az esetben a rendkívül szelektív elválasztási módszer és az érzékeny kimutatási módszer kombinációja a kromatográfiás módszerrel meghatározott anyagok körének észrevehető bővüléséhez vezet (nyomokban erősen mérgező anyagok, gyomirtó szerek, gyógyszerek, növekedésserkentők stb.).
A módszer részletei a szakirodalomban tisztázhatók.
Potenciometria– reverzibilis galvánelemek emf mérésén alapuló módszer az anyagok koncentrációjának meghatározására.
A gyakorlatban két analitikai módszert alkalmaznak: közvetlen potenciometria meghatározni a részecskeaktivitást, amely a galvánelem emf-jéből a Nernst-egyenlet segítségével számítható, ill. potenciometrikus titrálás , amelyben a kémiai anyagok aktivitásának változása a titrálási folyamat során a galvánelem emf-jének megváltozásához vezet.
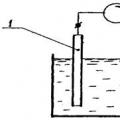
A potenciometrikus titrálás és a direkt potenciometria berendezése ugyanaz. A potenciometrikus mérőáramkör tartalmaz egy indikátorelektródát és egy stabil állandó potenciálú referenciaelektródát, valamint egy másodlagos eszközt. A módszer elvi diagramja a rizs. 3.

1 – indikátor elektróda; 2 - referencia elektróda
Rizs. 3. Potenciometrikus cella
Egy elektródapár potenciálja állandó. Az analit koncentrációjának megváltoztatása az oldatban megváltoztatja az áramkör EMF-jét. Az indikátorelektródák általában négyben vannak típusok, az alkalmazott membrántól függően, amely elválasztja az elektródoldatot a vizsgálati oldattól: 1) porszerű vagy kristályos anyagból készült homogén membránnal rendelkező elektródák; 2) heterogén membránnal rendelkező elektródák, amelyekben az elektróda hatóanyaga például szilikongumiban van elosztva; 3) folyékony membránnal ellátott elektródák, amelyekben a membrán egy semleges anyagra, például porózus üvegre felvitt oldat; 4) különböző kémiai összetételű üvegelektródák.
Az indikátorelektródák felveszik annak az oldatnak a potenciálját, amelybe helyezik őket. Van két kedves indikátor elektródák:
1) közömbös elektródák (nem roncsolhatók az elektrolízis során);
2) elektródák, amelyek a mérések során megváltoznak (oxidálódnak vagy redukálódnak).
Szerep közömbös elektródák(ezeket néha elektródáknak nevezik harmadik fajta) az, hogy elektronokat adjon vagy nyerjen, azaz. elektromos vezetők legyenek. Az ilyen elektródák készülhetnek aranyból, polírozott platinából, grafitból és egyéb anyagokból. Példák változó elektródákra (néha elektródáknak is nevezik) első fajta) lehetnek rézből, cinkből és más fémekből készült lemezek, valamint kinhidron és hidrogén indikátorelektródák. Ezen kívül indikátorelektródák lehetnek ion szelektív membránelektródák számos kation meghatározásához: Li +, Pb +, Cs +, Tl +, NH +, Na +, K +, Ag + stb. Referenciaelektródokként ( alapértelmezett elektródák), amelynek potenciálja a mérés során állandó marad, a leggyakrabban használt például a normál és decinormális kalomel (calomel) elektródák +0,282 V, illetve +0,334 V potenciállal, valamint a telített ezüst-klorid elektród +0,201 V potenciállal.
Ideális esetben egy galvánelem EMF közvetlen potenciometriás mérése a Nernst-egyenleten keresztül a meghatározandó részecske aktivitásához, vagy a koncentrációhoz köthető, ha ismertek a megfelelő aktivitási együtthatók:
![]()
Ahol E 0 – standard elektródpotenciál, V; R– gázállandó; T– abszolút hőmérséklet; F – Faraday-szám; n– elveszett vagy nyert elektronok száma; , [redukált] – oxidált, illetve redukált formák egyensúlyi koncentrációi, mol/dm 3 .
Ha behelyettesítjük az állandók referenciaértékeit, és a természetes logaritmusról a decimálisra lépünk, akkor 25 °C-os hőmérsékletre kapjuk:

A környezet állapotának jellemzésében a legfontosabb mutató e környezet pH-értéke, melynek meghatározása ( pH-metria ) jelenleg általában üveg indikátor (mérő) elektródákkal történik. A hosszú távú mérésekhez speciális kialakítású üvegelektródákat fejlesztettek ki kiegészítő eszközökkel, amelyek biztosítják az üvegmembrán tisztítását. Az elektrolit fóliával ellátott, félig áteresztő membránnal borított üvegelektródák különböző típusú szondák alapjául is szolgálnak ( érzékelők ), számos szennyező anyag (NH 3, CO 2, NO x, SO 2, H 2 S stb.) termelési körülményei között a víz és a levegő elemzésére használják.
Az ion-szelektív elektródák (ISE) létrehozásának folyamata lehetővé teszi az F – , I – , Br – , Cl – , CN – , SCN – , NO 3 – , NO 2 – , ClO 4 – , S 2 ionok monitorozását. – , Na + , A K + Ca 2+, Ag +, Cu 2+, Cd 2+, Pb 2+ koncentrációja 10 –2 és 10 –7 mol/l (körülbelül 1-10 –5 mg/ml) között van. Az ISE segítségével történő monitorozást gyorsaság, egyszerűség és nagyobb lehetőségek jellemzik a folyamatos mérések elvégzésére. Olyan ISE-ket fejlesztettek ki, amelyek szelektívek a szerves anyagok széles osztályára, valamint tömegükben izomerekre, felületaktív anyagokra és detergensekre, amelyek a termelési terület levegőjében és az ipari vállalkozások vízgazdálkodási rendszerében találhatók.
A potenciometriát a vízben lévő különféle redox (O/R) rendszerek redoxpotenciáljának mérésére is használják. A mérési eredmények általában vegyes potenciálnak felelnek meg, mivel a vízben általában több O/W rendszer létezik egyidejűleg.
Megjegyzendő, hogy a félvezető fémoxid kémiailag szelektív és ionszelektív térhatású tranzisztorokon (HSFT, ISFT) alapuló érzékelők alkalmazása ígéretes. Ezekben a rendszerekben a szelektivitást a membrán összetételének és a tranzisztor kapun lerakott rétegének megválasztásával érik el. A rendszer belemerül a vizsgált oldatba, és a referenciaelektróda és a tranzisztor kapuja közötti potenciálkülönbség modulálja a forrása és a lefolyó között folyó áramot. A membrán vagy a lerakódott réteg szelektivitása miatt a modulált áram az oldat megfelelő komponensének aktivitásának függvényévé válik. A félvezető érzékelők képezik a különféle gázok és gőzök monitorainak és elemzőinek alapját. Az ilyen érzékelők kis mérete lehetővé teszi, hogy egyetlen hordozón mozaik formájában egyesítsék őket, így olyan analizátort kapnak, amely a káros anyagok egész sorát képes figyelni. A mozaikban lévő egyes érzékelőktől érkező jeleket az analitikai rendszer mérőközpontja szekvenciálisan és periodikusan rögzítheti.
A mikroelektronika fejlődése lehetővé teszi kompakt szonda típusú analizátorok tervezését modern ISE-k segítségével. Ebben az esetben a szonda fogantyújába szerelhető egy áramkör, amely feldolgozza a környezeti vezérlőobjektum válaszát, sőt egy kijelző is.
A szakirodalomban megismerheti a módszer részleteit, , , .
Kulonometrikus az elemzési módszer az elektród reakció áramának mérése, amelybe a vizsgált anyag az elemzett áramlással belép a coulometriás cellába. A coulometriás cella sematikus diagramja látható rizs. 4.

1 – katódkamra; 2 – anódkamra; 3 – mikroampermérő
Rizs. 4. A coulometriás cella vázlata
A coulometriás elemzés azon alapul, hogy egy adott mintában egy adott elektrokémiai folyamat kvantitatív végrehajtására fordított villamos energia mennyiségét mérik, azaz egy adott mintában. feltéve, hogy az áram hatásfoka 100%. Ez az a villamosenergia-mennyiség a mérőcellával sorba kapcsolt áram-idő integrátor, vagy kulométer-elektrolizátor segítségével, amelyben százszázalékos áramhatékonysággal elektrokémiai folyamatot hajtanak végre, amelyhez egy áramkibocsátás társul. anyag, amelynek mennyisége könnyen és pontosan visszaállítható.
Vminek megfelelően Faraday törvénye:
m( x)/M(x) = m(k)/M(k),
Ahol m(x), m(k) – a meghatározandó anyag tömege xés a coulométerben felszabaduló anyag; M(x), M(k) – anyagegyenértékek moláris tömege xés a coulométerben felszabaduló anyag, g/mol.

A számítás elvégezhető a Faraday-törvényt leíró egyenlettel is:
![]()
ha az elemzés során áramerősséget mérnek én, A és az idő t, s, amelyet az elektrokémiai folyamat végrehajtására fordítottak.
Ennek a módszernek egy másik módosításában az ún
coulometriás titrálás
, a titráló elektrolitikus úton keletkezik a vizsgált oldatban adott áramerősség mellett. Az analitikai reakcióban a titrálószer fogyasztását az oldatban átáramló töltéssel helyettesítjük, amikor a titrálószer keletkezik, amíg el nem érjük az ekvivalenciapontot.
Az egyik A coulometriás módszerek előnyei az, hogy a titráló szabványosítási folyamat gyakran nem szükséges, mivel a számítások Faraday-állandón alapulnak, azaz a módszer abszolút, és lehetővé teszi a meghatározandó anyag mennyiségének becslését, nem pedig a koncentrációját. Az adott potenciállal rendelkező coulometria hátránya az elemzési eljárás időtartama, amely az elektrolízis teljes befejezésének szükségességéhez kapcsolódik. A számítástechnika lehetővé teszi ennek az időnek a csökkentését az elektrolízis végének előrejelzésével, az elektrolízis kezdeti szakaszára vonatkozó áram-idő görbe matematikai feldolgozásával, valamint az elektromosság mennyiségének vagy az oldatban lévő anyag koncentrációjának kiszámításával. Többkomponensű minták elemzésénél használható pásztázó coulometria , amelyben az elektrolízispotenciál folyamatosan vagy lépcsőzetesen változik. Az ilyen rendszerekben a coulometriás titrálás előnyösebb, mint a közvetlen kulometria, mivel a titráló reagens és a munkaközeg összetételének helyes megválasztásával könnyen elérhető a 100%-os áramhatékonyság a titrálószer előállításában. A coulometriás titrálás 0,01 és 100 mg közötti (néha 1 μg alatti) anyagok meghatározására alkalmazható. A munkaminta térfogata általában 10-50 ml. A módszert nagy pontosság jellemzi, a relatív hiba mikrogramm-tartalmak coulometriás titrálásával sem haladja meg a több tized százalékot. Optimális körülmények között a titrálás nagyon alacsony, 0,01%-os (rel.) általános hibával végezhető el. Különféle sav-bázis, redox; A kicsapás és a komplexometriás titrálási lehetőségek kulonometrikusan is végrehajthatók.
Kulonometrikus gázelemzőket és vízelemző készülékeket ("coulométer") fejlesztettek ki és gyártottak kén-dioxid és kénhidrogén (szulfátok és szulfidok), ózon (és hidrogén-peroxid), levegőben lévő klór (és aktív klór vízben) meghatározására, szén-monoxid és nitrogén-dioxid a levegőben (nitrátok és nitritek a vízben). A coulometriát elektrokémiai kimutatási eszközként is használják a folyadékkromatográfiában.
A módszer részletei a szakirodalomban találhatók.
Konduktometrikus módszer az elemzés az oldat elektromos vezetőképességének mérésén alapul. A konduktometrikus elemzési módszer abból áll, hogy megmérik az elektrolit oldat ellenállásának változását, amikor a keverék egy komponense felszívódik. A konduktometrikus berendezéseket például szén-monoxid és -dioxid, benzingőz, ammónia és mások meghatározására használják.
Az elektromos vezetőképesség az ellenállás reciproka R, mérete cm (Siemens), azaz. æ = 1/ R.
Egy oldat elektromos vezetőképessége az oldat térfogategységére jutó ionok számától függ, i.e. a koncentrációról VAL VEL, ezen ionok mobilitásáról – V. Ismert kapcsolatok alapján
![]()
Ahol Z– az elektródák közötti távolság; S – elektróda terület; k– arányossági együttható.
Egy adott elektródapárhoz, amelyek között állandó távolság van S/Z= konst. Akkor
![]() ,
,
Ahol k 1 = k(S/Z).
A konduktometriai számítások során az „elektromos vezetőképesség” æ 0 fogalmát használják:
![]()
A számítások során célszerű az ekvivalens elektromos vezetőképességet használni, amely egyenlő:
Ahol P - mólegyenértékek száma 1 cm 3 oldatban. Az ekvivalens elektromos vezetőképesség l ¥ végtelen hígításnál egyenlő a kationmozgások összegével Ués anion V.
Egy gyenge elektrolit oldat ekvivalens elektromos vezetőképességének és ennek az elektrolitnak a végtelen hígítású ekvivalens elektromos vezetőképességéhez viszonyított aránya egyenlő ennek az elektrolitnak a disszociációs fokával:
Nem specifikussága ellenére ezt a módszert más elektrokémiai módszerekkel összehasonlítva meglehetősen gyakran alkalmazzák környezeti monitoring rendszerekben. Ez azzal magyarázható, hogy a szennyezés, például a víz és a légkör értékelésénél nem fokozatos, hanem az ipari folyamatok kimeneti (végső) szabályozása lehetséges. A víz rendkívül alacsony elektromos vezetőképessége miatt gyakran elegendő a teljes szennyezőanyag-tartalom becslése, amit a konduktometria biztosít. A konduktometrikus módszerek környezetmonitoringban történő alkalmazásának jellemző példái a szennyvízben lévő tisztítószerek, az öntözőrendszerek szintetikus komponenseinek koncentrációja, valamint az ivóvíz minősége (sótartalma) elemzői. A konduktometrikus analizátorokat a levegő- és csapadékszennyező anyagok, például a SO 2 és a H 2 SO 4 folyamatos monitorozására használják. Továbbá közvetlen konduktometria bizonyos típusú szennyezések meghatározására használható közvetett módszerek, amelyek nagyon hatékonyan becsülik meg a fent felsorolt anyagok tartalmát, amelyek mérés előtt kölcsönhatásba lépnek speciálisan kiválasztott reagensekkel, és a rögzített elektromos vezetőképesség változást csak a megfelelő termékek jelenléte okozza a reakcióban. Így meghatározhatja a nitrogén-oxidokat az előammónia katalitikus redukciója után, valamint a HCl-t, a HBr-t és a CO 2-t Ba(OH) 2 -vel vagy NaOH-val végzett előzetes reakció után. A CO 2 meghatározásának leírt elve a vízben lévő szerves anyagok közvetett meghatározására is alkalmazható.
A klasszikus konduktometria mellett létezik egy nagyfrekvenciás változat is ( oszcillometria ), amelyben az indikátorelektródarendszer nem érintkezik a mintával. Ezt az elvet gyakran alkalmazzák a folyamatos vezetőképesség-elemzőkben.
Az elektrokémiai elemzési módszereket számos oktatási és speciális publikáció is ismerteti.
IRODALOM
1. Drugov Yu.S., Rodin A.A.Környezetelemző kémia.
Szentpétervár: 2002. – 464 p.
2. Pashkevich M.A., Shuisky V.F. Környezeti megfigyelés. oktatóanyag. Szentpétervári Állami Egyetem. – Szentpétervár, 2002. – 90 p.
3. Cattrall Robert W. Kémiai érzékelők. M.: Tudományos világ, 2000. – 144 p.
4. Turyan Ya.I., Ruvinsky O.E., Zaitsev P.M.Polarográfiai katalimetria. M.: Kémia, 1998. – 272 p.
5. Budnikov G.K., Maistrenko V.N., Murinov Yu.I. Voltammemetria módosított és ultramikroelektródákkal. M.: Nauka, 1994. – 239 p.
6. Brainina Kh.Z., Neiman E.Ya., Slepushkin V.V. Inverziós elektroanalitikai módszerek. M.: 1988. – 240 p.
7. Salikhdzhanova R.F. satöbbi. Polarográfok és felhasználásuk a gyakorlati elemzésben és kutatásban. M.: Kémia, 1988. – 192 p.
8. Kaplan B.Ya., Pats R.G., Salikhdzhanova R.F. AC voltammetria. M.: Kémia, 1985. – 264.
9. Bond A.M. Polarográfiai módszerek az analitikai kémiában. M.: Kémia, 1983.
10. Efremenko O.A. Potenciometriai elemzés. M.: MMA im. ŐKET. Sechenova, 1998.
11. Útmutató az ion-szelektív elektródák alkalmazásához. M.: Mir, 1986.
12. Koryta I. Ionok, elektródák, membránok. M.: Mir, 1983.
13. Nikolsky B.V., Materova E.A. Ion szelektív elektródák. L.: Kémia, 1980.
14. Efremenko O.A.Kulonometrikus titrálás. M.: MMA im. ŐKET. Sechenova, 1990.
15. Khudyakova T.A., Koreshkov A.P. Konduktometrikus elemzési módszer. Tankönyv egyetemek számára. M.: Felsőiskola, 1975. – 207 p.
16. Budnikov G.K., Maistrenko V.N., Vyaselev M.R. A modern elektromos elemzés alapjai. M.: Kémia, 2000.
17. Prokhorova G.V. Bevezetés az elektrokémiai elemzési módszerekbe. M.: Moszkvai Állami Egyetemi Kiadó, 1991. – 97 p.
18. Elektroanalitikai módszerek a környezeti monitoringban. /Szerk. R. Kalvoda, R. Zyka, K. Shtulik és mások M.: Kémia, 1990. – 240 p.
19. Plambeck J.Elektrokémiai elemzési módszerek. Elméleti és alkalmazási alapismeretek./Ford. angolról M.: Mir, 1986.
Munkaleírás
A modern termelési ágak és az emberek társadalmi élete sajátos feladatokat ró a termékminőség-ellenőrzés fizikai és kémiai elemzési módszereire. Az egyik fő fizikai-kémiai elemzési módszer az elektrokémiai elemzési módszerek.
Ezekkel a módszerekkel gyorsan és meglehetősen pontosan meg lehet határozni számos termékminőségi mutatót.
Az anyagok összetételének elemzésére szolgáló elektrokémiai módszereket széles körben alkalmazzák a különböző iparágakban. Lehetővé teszik a termékminőségre vonatkozó eredmények automatizálását és a jogsértések kijavítását a gyártás leállítása nélkül. Az élelmiszeriparban ezek a módszerek meghatározzák a termék sav-bázis egyensúlyát, a káros és mérgező anyagok jelenlétét és egyéb mutatókat, amelyek nemcsak az élelmiszerek minőségét, hanem biztonságát is befolyásolják.
Az elektrokémiai elemzésre tervezett berendezések viszonylag olcsók, hozzáférhetőek és könnyen használhatók. Ezért ezeket a módszereket széles körben alkalmazzák nemcsak a speciális laboratóriumokban, hanem számos iparágban is.
E tekintetben a célja ennek a ku
BEVEZETÉS 2
ELMÉLETI RÉSZ 3
1.1 A fizikai-kémiai elemzési módszerek általános jellemzői 3
1.2 Az elektrokémiai módszerek jellemzői 4
1.3 Az elektrokémiai elemzési módszerek osztályozása 5
2 KÍSÉRLETI-GYAKORLATI RÉSZ 15
KÖVETKEZTETÉS 21
IRODALOM 22
Bevezetés
1. fejezet Általános fogalmak. Az elektrokémiai elemzési módszerek osztályozása
2. fejezet Potenciometrikus elemzési módszerek (potenciometria)
1 A módszer elve
3 Potenciometrikus titrálás
3. fejezet Konduktometrikus elemzési módszer
1 A módszer elve. Alapfogalmak
2 A konduktometria elve
3 Konduktometrikus titrálás
4. fejezet Konduktometriai elemzés (conduktometria)
1 A módszer lényege
2 Kvantitatív polarográfiai elemzés
3 A polarográfia alkalmazásai
5. fejezet Amperometrikus titrálás
6. fejezet Kulometriás elemzés (kulometria)
1 A módszer elve
3 Kulonometrikus titrálás
Következtetés
Bibliográfia
BEVEZETÉS
Az elektrokémiai elemzési módszerek a kvalitatív és kvantitatív elemzési módszerek összessége, amelyek a vizsgált közegben vagy a határfelületen előforduló elektrokémiai jelenségeken alapulnak, és az analit szerkezetének, kémiai összetételének vagy koncentrációjának változásaihoz kapcsolódnak.
Az elektrokémiai elemzési módszerek öt fő csoportra oszthatók: potenciometria, voltammetria, kulometria, konduktometria és amperometria.
Ezeknek a módszereknek a kvantitatív elemzésben való alkalmazása az elektrokémiai folyamat során mért paraméterek értékének az elektrokémiai folyamatban részt vevő elemzett oldatban lévő elválasztott anyagtól való függésén alapul. Ilyen paraméterek közé tartozik az elektromos potenciál különbsége és az elektromosság mennyisége. Az elektrokémiai folyamatok olyan folyamatok, amelyek egyidejűleg járnak együtt kémiai reakcióval és a rendszer elektromos tulajdonságainak megváltozásával, amelyet ilyen esetekben elektrokémiai rendszernek nevezhetünk. Az analitikai gyakorlatban egy elektrokémiai rendszer jellemzően egy elektrokémiai cellát tartalmaz, amely egy elektromosan vezető vizsgálati oldatot tartalmazó edényt tartalmaz, amelybe elektródákat merítenek.
Vannak direkt és közvetett elektrokémiai módszerek. A direkt módszereknél az áramerősség (potenciál stb.) függését a meghatározandó komponens koncentrációjától használják. Az indirekt módszereknél az áramerősséget (potenciált stb.) mérik, hogy megtalálják a meghatározandó komponens titrálásának végpontját megfelelő titrálóval, vagyis a mért paraméter titráló térfogattól való függését használják.
1. FEJEZET ÁLTALÁNOS FOGALMAK. AZ ELEKTROKÉMIAI ANALÍZIS MÓDSZEREK OSZTÁLYOZÁSA
Az elektroanalitikai kémia magában foglalja az elektrokémiai elemzési módszereket, amelyek az elektród reakciókon és az elektromosság oldatokon keresztül történő átvitelén alapulnak.
Az elektrokémiai módszerek alkalmazása a kvantitatív elemzésben az elektrokémiai folyamatok mért paraméterei (elektromos potenciálkülönbség, áramerősség, villamos energia mennyisége) értékeinek a vizsgált oldatban lévő analit tartalmától való függésének a felhasználásán alapul. ezt az elektrokémiai folyamatot. Az elektrokémiai folyamatok olyan folyamatok, amelyek egyidejűleg kémiai reakciók végbemenetelével és a rendszer elektromos tulajdonságainak megváltozásával járnak, amit ilyen esetekben elektrokémiai rendszernek nevezhetünk. Az analitikai gyakorlatban egy elektrokémiai rendszer általában tartalmaz egy elektrokémiai cellát, beleértve egy elektromosan vezető vizsgálati oldattal ellátott edényt, amelybe elektródákat merítenek.
Az elektrokémiai elemzési módszerek osztályozása. Az elektrokémiai elemzési módszereket többféleképpen osztályozzák, az osztályozás azon alapul, hogy figyelembe veszik a rendszerben lévő elektromos energiaforrás jellegét. A módszereknek két csoportja van:
a) Módszerek külső (külső) potenciál kiváltása nélkül.
Az elektromos energia forrása maga az elektrokémiai rendszer, amely egy galvánelem (galvanikus áramkör). E módszerek közé tartoznak a potenciometrikus módszerek. Az elektromotoros erő - EMF - és az elektródpotenciálok egy ilyen rendszerben az oldatban lévő analit tartalmától függenek.
b) Módszerek külső (külső) potenciál rákényszerítésével. Ezek a módszerek a következők:
konduktometrikus elemzés - az oldatok elektromos vezetőképességének mérésén alapul, koncentrációjuk függvényében;
voltammetriás analízis - az alkalmazott ismert potenciálkülönbség és az oldat koncentrációjának függvényében áram mérésén alapul;
coulometriás elemzés - az oldaton áthaladó elektromosság mennyiségének mérésén alapul, annak koncentrációjának függvényében;
elektrogravimetriás elemzés - elektrokémiai reakció termékének tömegének mérésén alapul.
Osztályozás az elektrokémiai módszerek alkalmazási módja szerint. Vannak közvetlen és közvetett módszerek.
a) Közvetlen módszerek. Az elektrokémiai paramétert az oldat koncentrációjának ismert függvényeként mérjük, és a megfelelő mérőeszköz leolvasása alapján meghatározzuk az oldatban a meghatározandó anyag tartalmát.
b) A közvetett módszerek olyan titrálási módszerek, amelyeknél a titrálás végét a rendszer elektromos paramétereinek mérése alapján határozzák meg.
Ennek az osztályozásnak megfelelően megkülönböztetik például a közvetlen konduktometriát és a konduktometrikus titrálást.
2. FEJEZET. POTENCIOMETRIAI ELEMZÉSI MÓDSZER (POTENCIOMETRIA)
1 A módszer elve
A potenciometriai analízis (potenciometria) az emf és az elektródpotenciálok mérésén alapul, a vizsgált oldat koncentrációjának függvényében.
Ha egy elektrokémiai rendszerben - galvanikus cellában - reakció lép fel az elektródákon:
aA+bB↔dD + eE
n elektron átvitelével, akkor ennek a reakciónak az emf E Nernst-egyenlete a következő:
E꞊E˚- RTnFlnaDda Eea(A)a aBb
ahol szokásos módon E° a reakció standard EMF-je (a standard elektródpotenciálok különbsége), R a gázállandó, T a reakció végbemenetelének abszolút hőmérséklete, F a Faraday-szám; a(A), a(B), a(D) és i(E) - a reakcióban részt vevő reagensek aktivitása. A (10.1) egyenlet egy reverzibilisen működő galvánelem emf-ére érvényes.
Szobahőmérséklet esetén a (10.1) egyenlet a következő formában ábrázolható:
E꞊E˚- 0,059nlnaDda Eea(A)a aBb
Olyan körülmények között, ahol a reagensek aktivitása megközelítőleg megegyezik koncentrációjukkal, az (1) egyenletből a (3) egyenlet lesz:
꞊E˚- RTnFlncDdc EecAa aBb
ahol c(A), c(B), c(E), c(D) a reagensek koncentrációja. Szobahőmérséklet esetén ez az egyenlet a következőképpen ábrázolható: (4):
꞊E˚- 0,059nlncDdc EecAa aBb
A potenciometrikus mérésekhez egy elektrokémiai cellában két elektródát használnak: egy indikátorelektródát, amelynek potenciálja a vizsgált oldatban lévő analit (potenciálmeghatározó) anyag koncentrációjától függ, és egy referenciaelektródát, amelynek potenciálja állandó marad. elemzési körülmények között. Ezért az EMF (1)-(4) egyenletekkel meghatározott nagysága e két elektróda valós potenciálja közötti különbségként számítható ki.
A potenciometriában a következő típusú elektródákat alkalmazzák: első, második típusú elektródák, redox, membránelektródák.
Az első típusú elektródák olyan elektródák, amelyek az elektróda anyagában közös kation segítségével megfordíthatók. Az első típusú elektródák három típusa létezik.
a) Ugyanazon fém sójának oldatába merített M fém. Az ilyen elektródák felületén reverzibilis reakció lép fel:
Mn++ne = M
Egy ilyen első típusú elektród valós potenciálja a fémkationok a(Mn+) aktivitásától függ, és az (5)-(8) egyenletek írják le.
Általában bármilyen hőmérsékletre:
꞊E˚+ RTnFln a(Mn+)
Szobahőmérséklethez:
꞊E˚+ 0,059nln a(Mn+)
Alacsony c(Mn+) koncentrációnál, amikor az a(Mn+) fémkationok aktivitása megközelítőleg megegyezik koncentrációjukkal:
꞊E˚+ RTnFln c(Mn+)
Szobahőmérséklethez:
b) Gázelektródák, például hidrogénelektródák, beleértve a szabványos hidrogénelektródát is. A reverzibilisen működő gáz-hidrogénelektród potenciálját a hidrogénionok aktivitása határozza meg, pl. az oldat pH-értéke és szobahőmérsékleten egyenlő:
꞊E˚+ 0,059 lg a(H30+) = 0,059 lg a(H3O+) = -0,059 рН
mivel egy hidrogénelektród esetében a standardpotenciál értéke nulla ( £° =0), és az elektród reakciójának megfelelően: H++e = N a reakcióban részt vevő elektronok száma eggyel egyenlő: n = 1. c) Amalgám elektródák, amelyek ugyanazon fém kationjait tartalmazó oldatba merített fém-amalgám. Az első típusú ilyen elektródák potenciálja az a(Mn+) fémkationok oldatban lévő aktivitásától és az a(M) fém aktivitásától függ az amalgámban: ꞊E˚+ RTnFlna(Mn+)a(M) Az amalgámelektródák nagymértékben reverzibilisek. A második típusú elektródák anionos reverzibilisek. A második típusú elektródák következő típusait különböztetjük meg. a) Fém, amelynek felülete ugyanannak a fémnek gyengén oldódó sójával van bevonva, és olyan oldatba merítve, amely a nehezen oldódó sót alkotó anionokat tartalmazza. Ilyen például az Ag|AgCl, KS1 ezüst-klorid elektród vagy a Hg|Hg2Cl2, KS1 kalomelelektród. Az ezüst-klorid elektróda egy vízben gyengén oldódó sóval, AgCI-vel bevont ezüsthuzalból áll, amelyet kálium-klorid vizes oldatába merítenek. Reverzibilis reakció megy végbe az ezüst-klorid elektródán A kalomelelektróda fémhiganyból áll, amelyet rosszul oldódó higany(1)-klorid, Hg2Cl2-kalomel pasztával vonnak be, és kálium-klorid vizes oldatával érintkezik. Reverzibilis reakció megy végbe a kalomel elektródán: Cl2 + 2e = 2Hg + 2SG. A második típusú elektródák valós potenciálja az anionok aktivitásától és egy reverzibilis elektródától függ, amelyen a reakció végbemegy: Ne = M + An- a (9)-(12) Nernst-egyenletek írják le. Általában bármely elfogadható hőmérsékleten T: ꞊E˚- RTnFln a(An-) Szobahőmérséklethez: ꞊E˚- 0,059nln a(An-) Olyan körülmények között, amelyekben az anionok aktivitása megközelítőleg megegyezik c(A"~) koncentrációjukkal: E꞊E˚- RTnFln c(An-) Szobahőmérséklethez: ꞊E˚- 0,059nln c(An-) Például az ezüst-klorid és kalomel elektródák E1 és E2 valós potenciálja szobahőmérsékleten a következőképpen ábrázolható: ꞊E1˚- 0,0591 g a(Cl-), ꞊E2˚- 0,0591 g a(Cl-). A második típusú elektródák nagymértékben reverzibilisek és stabil működésűek, ezért gyakran használják referenciaelektródákként, amelyek képesek stabilan fenntartani az állandó potenciálértéket. b) Második típusú gázelektródák, például klórelektród Pt, Cl2 KS1. A második típusú gázelektródákat ritkán használják kvantitatív potenciometrikus elemzésben. A redox elektródák inert anyagból (platina, arany, volfrám, titán, grafit stb.) állnak, amelyeket oxidált oxot és ennek redukált vörös formáit tartalmazó oldatba merítenek. Kétféle redox elektróda létezik: a) elektródák, amelyek potenciálja nem függ a hidrogénionok aktivitásától, például Pt | FeCl3, FeCl2, Pt | K3, K4 stb.; b) elektródák, amelyek potenciálja a hidrogénionok aktivitásától függ, például kinhidron elektród. A redox elektródán, amelynek potenciálja nem függ a hidrogénionok aktivitásától, reverzibilis reakció megy végbe: Ox + ne = Piros Egy ilyen redox elektród valós potenciálja az adott anyag oxidált és redukált formáinak aktivitásától függ, és egy reverzibilisen működő elektródát a körülményektől függően (a fentebb tárgyalt potenciálokkal analóg módon) a Nernst-egyenletek írják le ( 13)-(16): ꞊E˚+ RTnFln a (Ox)a (piros)꞊E˚+ 0.059nlg a (Ox)a (piros)꞊E˚+ RTnFln c(Ox)c (piros)꞊E˚+ 0.059nlg c (Ox) c (piros) Ha hidrogénionok vesznek részt az elektród reakciójában, akkor aktivitásukat (koncentrációjukat) a megfelelő Nernst-egyenletekben minden konkrét esetben figyelembe veszik. A membrán vagy ion-szelektív elektródák olyan elektródák, amelyek reverzibilisek bizonyos szilárd vagy folyékony membrán által szorbeált ionok (kationok vagy anionok) számára. Az ilyen elektródák valós potenciálja a membrán által szorbeált oldatban lévő ionok aktivitásától függ. A szilárd membránelektródák egy nagyon vékony membránt tartalmaznak, melynek mindkét oldalán különböző, ugyanazokat a meghatározandó ionokat tartalmazó, de eltérő koncentrációjú oldatok találhatók: egy oldat (standard) a meghatározandó ionok pontosan ismert koncentrációjával, ill. a meghatározandó ionok ismeretlen koncentrációjával. A két oldat eltérő ionkoncentrációja miatt a membrán különböző oldalán lévő ionok egyenlőtlen mennyiségben szorbeálódnak, és a membrán különböző oldalain lévő ionok szorpciójából származó elektromos töltés is eltérő. Ennek eredményeként membránpotenciál-különbség keletkezik. Az ionok membránion-szelektív elektródákkal történő meghatározását ionometriának nevezzük. Mint fentebb említettük, a potenciometrikus méréseknél az elektrokémiai cella két elektródát tartalmaz - egy indikátorelektródát és egy referenciaelektródát. A cellában generált EMF nagysága megegyezik a két elektróda közötti potenciálkülönbséggel. Mivel a referenciaelektróda potenciálja a potenciometrikus meghatározás körülményei között állandó marad, az EMF csak az indikátorelektród potenciáljától függ, pl. egyes ionok oldatban lévő aktivitásáról (koncentrációjáról). Ez az alapja egy adott anyag koncentrációjának potenciometrikus meghatározásának a vizsgált oldatban. Az oldatban lévő anyag koncentrációjának potenciometrikus meghatározásához közvetlen potenciometriát és potenciometrikus titrálást is alkalmaznak, bár a második módszert sokkal gyakrabban használják, mint az elsőt. Egy anyag koncentrációjának közvetlen potenciometriával történő meghatározását általában a kalibrációs görbe módszerével vagy a standard addíciós módszerrel végzik. a) Kalibrációs gráf módszer. Készítsen 5-7 standard oldatból álló sorozatot ismert analittartalommal. A standard oldatokban az analit koncentrációja és az ionerősség nem térhet el nagymértékben a vizsgált oldat koncentrációjától és ionerősségétől: ilyen körülmények között a meghatározási hibák csökkennek. Az összes oldat ionerősségét közömbös elektrolit bevezetésével állandó értéken tartják. A standard oldatokat egymás után bevezetjük egy elektrokémiai (potenciometrikus) cellába. Ez a cella jellemzően egy üvegpohár, amelybe egy indikátorelektródát és egy referenciaelektródát helyeznek el. A standard oldatok EMF-jét úgy mérjük, hogy az elektródákat és az üveget desztillált vízzel alaposan mossuk, mielőtt a cellát minden standard oldattal megtöltjük. A kapott adatok alapján EMF-log c koordinátákban kalibrációs grafikont készítünk, ahol c az analit koncentrációja a standard oldatban. Ez a grafikon általában egy egyenes. Ezután az elemzett oldatot az elektrokémiai cellába töltjük (a cella desztillált vízzel történő mosása után), és megmérjük a cella emf-jét. A kalibrációs grafikon segítségével log c(X) található, ahol c(X) az analit koncentrációja a vizsgált oldatban. b) Standard összeadási módszer. A c(X) koncentrációjú elemzett oldat ismert V(X) térfogatát az elektrokémiai cellába adjuk, és megmérjük a cella emf-jét. Ezután ugyanahhoz az oldathoz adunk egy pontosan mért kis térfogatú V(st) standard oldatot ismert, kellően nagy koncentrációjú c(st) analittal, és ismét meghatározzuk a cella emf-jét. Számítsuk ki az analit c(X) koncentrációját a vizsgált oldatban a (10.17) képlet segítségével: c(X)= c(st) V (st)V X+ V (st) Ahol △ E két mért EMF-érték különbsége, n az elektródreakcióban részt vevő elektronok száma. Közvetlen potenciometria alkalmazása. A módszer a hidrogénionok (az oldatok pH-ja), az anionok és a fémionok koncentrációjának meghatározására szolgál (ionometria). A direkt potenciometria alkalmazásakor fontos szerepet játszik a megfelelő indikátorelektróda kiválasztása és az egyensúlyi potenciál pontos mérése. Az oldatok pH-értékének meghatározásakor elektródákat használnak indikátorelektródákként, amelyek potenciálja a hidrogénionok koncentrációjától függ: üveg, hidrogén, kinhidron és mások. Gyakrabban használnak hidrogénionokban reverzibilis membránüveg elektródát. Egy ilyen üvegelektróda potenciálját a hidrogénionok koncentrációja határozza meg, ezért az üvegelektródát indikátorként tartalmazó áramkör EMF-jét szobahőmérsékleten a következő egyenlet írja le: K + 0,059 рН, ahol a K állandó a membrán anyagától és a referenciaelektróda jellegétől függ. Az üvegelektróda lehetővé teszi a pH meghatározását a pH = 0-10 tartományban (gyakrabban a pH = 2-10 tartományban), és nagyon reverzibilis és működés közben stabil. A régebben gyakran használt kinhidron elektród egy redox elektród, amelynek potenciálja a hidrogénionok koncentrációjától függ. Egy platinahuzalból áll, amelyet kinhidronnal telített savas oldatba (általában HC1) merítenek, kinon és hidrokinon ekvimolekuláris vegyülete C6H402 összetételű. C6H4(OH)2 (sötétzöld por, vízben gyengén oldódik). A kinhidron elektród vázlatos jelölése: Pt | kinhidron, HC1. Redox reakció megy végbe a kinhidron elektródán: C6H402 + 2H+ + 2e = C6H4(OH)2 A kinhidron elektród potenciálját szobahőmérsékleten a képlet írja le E°-0,059рН. A kinhidron elektróda lehetővé teszi az oldatok pH-értékének mérését a pH = 0-8,5 tartományban. pH-n< 0 хингидрон гидролитически расщепляется: при рН >A 8,5 hidrokinon, amely egy gyenge sav, semlegesítési reakción megy keresztül, erős oxidáló és redukáló szerek jelenlétében a kinhidron elektróda nem használható. A membránion-szelektív elektródákat, amint fentebb említettük, az ionometriában indikátorként használják különböző kationok (Li+, Na+, K+ Mg2t, Ca2+, Cd2+, Fe2+, Ni2+ stb.) ionok (F-, Cl-, Br -, I-, S2- stb.). A közvetlen potenciometria előnyei közé tartozik a mérések egyszerűsége és gyorsasága, a mérésekhez kis mennyiségű megoldásra van szükség. 3 Poteniometrikus titrálás A potenciometrikus titrálás a vizsgált oldatban lévő analit titrálására fordított titrálószer térfogatának meghatározására szolgáló módszer az EMF mérésével (a titrálási folyamat során) indikátorelektródából álló galvánkör segítségével. és egy referenciaelektródát. A potenciometrikus titrálás során az elektrokémiai cellában elhelyezett elemzett oldatot titráljuk megfelelő titráló, amely a titrálás végét a mért áramkör EMF-jének éles változásával rögzíti - az indikátorelektróda potenciálja, amely a megfelelő ionok koncentrációjától függ, és élesen változik az ekvivalencia ponton. Az indikátorelektróda potenciáljának változását a titrálási folyamat során a hozzáadott titrálószer térfogatától függően mérjük. A kapott adatok alapján felállítunk egy potenciometrikus titrálási görbét, és ebből a görbéből határozzuk meg az üzemanyagcellában elfogyasztott titrálószer térfogatát. A potenciometrikus titráláshoz nincs szükség a tüzelőanyag-elem közelében színváltoztató indikátorok használatára. Potenciometrikus titrálás alkalmazása. A módszer univerzális, minden típusú titrálásnál használható a titrálás befejezésének jelzésére: sav-bázis, redox, komplexmetriás, precipitációs és nem vizes közegben végzett titráláskor. Üveg-, higany-, ion-szelektív, platina- és ezüstelektródákat használnak indikátorelektródákként, referenciaelektródákként pedig kalomelt, ezüst-kloridot és üvegelektródákat. A módszer nagy pontossággal és nagy érzékenységgel rendelkezik: lehetővé teszi a titrálást zavaros, színes, nem vizes közegben, valamint a keverék komponenseinek külön-külön történő meghatározását egy vizsgált oldatban, például a klorid- és jodidionok elkülönített meghatározását argentometriás titrálás során. A potenciometrikus titrálási módszereket számos gyógyászati anyag elemzésére használják, például aszkorbinsav, szulfa-gyógyszerek, barbiturátok, alkaloidok stb. A konduktometrikus elemzés megalapítójának F.V.G. német fizikust és fizikai kémikust tartják. Kohlrausch (1840-1910), aki először 1885-ben javasolt egy egyenletet, amely összefüggést állapít meg az erős elektrolitok oldatainak elektromos vezetőképessége és koncentrációja között. BAN BEN 40-es évek közepe XX század nagyfrekvenciás konduktometrikus titrálási módszert fejlesztettek ki. A 60-as évek eleje óta. XX század A konduktometriás detektorokat elkezdték használni a folyadékkromatográfiában. 1 A módszer elve. Alapfogalmak A konduktometriai analízis (conduktometria) az elektrolitoldatok elektromos vezetőképessége (elektromos vezetőképessége) és koncentrációjuk összefüggésének felhasználásán alapul. Az elektrolit oldatok - a második típusú vezetők - elektromos vezetőképességének megítélése az elektromos ellenállásuk elektrokémiai cellában történő mérése alapján történik, amely egy üvegedény (üveg), amelybe két elektródát forrasztottak, amelyek között a vizsgált elektrolit oldat található. A cellán váltakozó elektromos áram halad át. Az elektródák leggyakrabban fémplatinából készülnek, amelyet az elektródák felületének növelése érdekében szivacsos platinaréteggel vonnak be a platinavegyületek oldatokból történő elektrokémiai leválasztásával (platinizált platinaelektródák). Az elektrolízis és polarizáció folyamataival kapcsolatos szövődmények elkerülése érdekében a konduktometriás méréseket váltakozó elektromos térben végezzük. Az elektródák közötti elektrolitoldat réteg R elektromos ellenállása az első típusú vezetők elektromos ellenállásához hasonlóan egyenesen arányos ennek a rétegnek a hosszával (vastagságával) l és fordítottan arányos az elektródák S felületével: R= ρ lS lkS ahol a p arányossági együtthatót fajlagos elektromos ellenállásnak, a fordított k = 1/p értéket pedig fajlagos elektromos vezetőképességnek (elektromos vezetőképességnek) nevezzük. Mivel az R elektromos ellenállást ohmban mérjük, az elektrolit oldat réteg l vastagságát cm-ben, az elektródák felületét S cm2-ben adjuk meg, a k fajlagos elektromos vezetőképességet Ohm-1 egységekben mérjük. cm-1, vagy mivel Ohm-1 Siemens (Sm), akkor - Sm egységekben cm-1. Fizikai értelmében a fajlagos elektromos vezetőképesség egy 1 cm oldalhosszúságú kocka oldalai között elhelyezkedő elektrolitréteg elektromos vezetőképessége, amely számszerűen megegyezik az elektrolitoldat-rétegen áthaladó árammal, amelynek keresztmetszete 1 cm. 1 cm2, 1 V/cm alkalmazott elektromos potenciálgradiens mellett. A fajlagos elektromos vezetőképesség az elektrolit és az oldószer természetétől, az oldat koncentrációjától és a hőmérséklettől függ. Az elektrolitoldat növekvő koncentrációjával a fajlagos elektromos vezetőképessége először növekszik, majd maximumon halad át, majd csökken. Az elektromos vezetőképesség változásának ilyen jellege a következő okokra vezethető vissza. Kezdetben az elektrolitkoncentráció növekedésével az ionok - áramot szállító részecskék - száma megnő mind az erős, mind a gyenge elektrolitoknál. Ezért az oldat elektromos vezetőképessége (a rajta áthaladó elektromos áram) megnő. Majd az oldat koncentrációjának növekedésével megnő a viszkozitása (az ionok mozgási sebességének csökkentése) és az ionok közötti elektrosztatikus kölcsönhatások, ami megakadályozza az elektromos áram növekedését, és kellően nagy koncentráció esetén segíti annak csökkentését. A gyenge elektrolitok oldataiban a koncentráció növekedésével az elektrolit molekulák disszociációs foka csökken, ami az ionok - vezető részecskék - számának csökkenéséhez és a fajlagos elektromos vezetőképesség csökkenéséhez vezet. Erős elektrolitok nagy koncentrációjú oldataiban ionos asszociátumok (ionos ikrek, pólók stb.) képződése lehetséges, ami szintén kedvez az elektromos vezetőképesség csökkenésének. Az elektrolit oldatok fajlagos elektromos vezetőképessége a hőmérséklet emelkedésével növekszik az oldatok viszkozitásának csökkenése miatt, ami az ionok mozgási sebességének növekedéséhez, gyenge elektrolitoknál pedig ionizációs fokuk növekedéséhez vezet. (ionokká történő disszociáció). Ezért a kvantitatív konduktometrikus méréseket állandó hőmérsékleten, a konduktometrikus cella termosztálásával kell végezni. A fajlagos elektromos vezetőképesség mellett a konduktometria egyenértékű X elektromos vezetőképességet és p moláris elektromos vezetőképességet alkalmaz. Fizikai értelemben az ekvivalens elektromos vezetőképesség X az azonos elektródák között elhelyezkedő 1 cm vastag elektrolitoldatréteg elektromos vezetőképessége, amelynek területe olyan, hogy a közéjük zárt elektrolit oldat térfogata 1 g-ekvivalens oldott anyagot tartalmazzon. Ebben az esetben az ekvivalens moláris tömege az egységnyi töltésszámú azonos részecskék moláris tömege („töltés”), például H+, Br -, 12Ca2+, 13Fe3+ stb. Az ekvivalens elektromos vezetőképesség az elektrolitoldat koncentrációjának csökkenésével növekszik. Az ekvivalens elektromos vezetőképesség maximális értéke az oldat végtelen hígításával érhető el. Az egyenértékű elektromos vezetőképesség a fajlagos vezetőképességhez hasonlóan a hőmérséklet emelkedésével növekszik. Az X ekvivalens elektromos vezetőképesség a k fajlagos elektromos vezetőképességgel van összefüggésben (20): λ= 1000 kc A közvetlen konduktometria során a vizsgált oldatban lévő anyag koncentrációját az oldat fajlagos elektromos vezetőképességének mérési eredményei alapján határozzák meg. A mérési adatok feldolgozásakor két módszert alkalmaznak: a számítási módszert és a kalibrációs gráf módszert. Számítási módszer. A (10.20) egyenletnek megfelelően az oldatban lévő elektrolit ekvivalens c moláris koncentrációja kiszámítható, ha ismert a k fajlagos elektromos vezetőképesség és az ekvivalens elektromos vezetőképesség : c = 1000 kλ A fajlagos elektromos vezetőképesség meghatározása kísérleti úton történik egy termosztált konduktometrikus cella elektromos ellenállásának mérése alapján. Az oldat ekvivalens elektromos vezetőképessége λ egyenlő a kationmozgások összegével λ+ és X anion λ -:
λ = λ + + λ-
Ha a kation és az anion mobilitása ismert, akkor a koncentráció a (24) képlet segítségével számítható ki: c = 1000 kλ + + λ- Erre akkor kerül sor, ha direkt konduktometriával határozzuk meg a rosszul oldódó elektrolit koncentrációját telített oldatában (kalcium, bárium-szulfátok, ezüsthalogenidek stb.). Kalibrációs grafikon módszer. Egy sor standard oldatot készítenek, amelyek mindegyike pontosan ismert koncentrációjú analitot tartalmaz, és elektromos vezetőképességüket állandó hőmérsékleten, termosztált konduktometrikus cellában mérik. A kapott adatok alapján kalibrációs grafikont készítünk, amely az abszcissza tengelyen ábrázolja a standard oldatok koncentrációját, az ordináta tengely mentén pedig a fajlagos elektromos vezetőképesség értékeit. A (24) egyenletnek megfelelően az ábrázolt grafikon általában egy egyenest ábrázol a koncentrációváltozások viszonylag kis tartományában. Széles koncentrációtartományban, amikor a (24) egyenletben szereplő kation és anion mobilitása észrevehetően változhat, eltérések figyelhetők meg a lineáris függéstől. Ezután szigorúan azonos feltételek mellett megmérjük a vizsgált oldatban meghatározott, ismeretlen koncentrációjú, c(X) elektrolit fajlagos elektromos vezetőképességét, és a grafikonon megtaláljuk a kívánt c(X) értéket. Így határozzák meg például a báriumtartalmat baritvízben - bárium-hidroxid telített oldatában. Közvetlen konduktometria alkalmazása. A közvetlen konduktometriás módszert az egyszerűség és a nagy érzékenység jellemzi. A módszer azonban nem túl szelektív. A közvetlen konduktometriát korlátozottan használják az elemzésben. Alkalmas a rosszul oldódó elektrolitok oldhatóságának meghatározására, a desztillált víz és folyékony élelmiszerek (tej, italok stb.) minőségének ellenőrzésére, ásványi, tengeri, folyóvízi és egyéb esetekben az összes sótartalom meghatározására. . 3 Konduktometrikus titrálás A konduktometrikus titrálás során a titrálás előrehaladását a két (általában platinizált platinából készült) inert elektróda között elhelyezkedő konduktometrikus cellában elhelyezett elemzett oldat elektromos vezetőképességének változásai követik nyomon. A kapott adatok alapján konduktometrikus titrálási görbét rajzolunk, amely tükrözi a titrált oldat elektromos vezetőképességének a hozzáadott titrálószer térfogatától való függését. A titrálás végpontját leggyakrabban a titrálási görbe azon szakaszainak extrapolálásával találjuk meg, ahol a meredeksége változik, ilyenkor a TE közelében színváltoztató indikátorok használata nem szükséges. A konduktometrikus titrálás során különféle típusú reakciókat alkalmaznak: sav-bázis, redox, kicsapás, komplexképző folyamatok. Konduktometrikus titrálás alkalmazása. A konduktometrikus titrálási módszernek számos előnye van. A titrálás végezhető zavaros, színes, átlátszatlan közegben. A módszer érzékenysége meglehetősen magas - akár ~10~* mol/l; a meghatározási hiba 0,1 és 2% között van. Az elemzés automatizálható. A módszer hátrányai közé tartozik az alacsony szelektivitás. A nagyfrekvenciás (rádiófrekvenciás) konduktometrikus titrálás fogalma. A titrálás előrehaladását módosított váltóáramú konduktometrikus technikával követik nyomon, amelyben a váltakozó áram frekvenciája elérheti a másodpercenkénti nagyságrendű oszcillációt. Az elektródákat jellemzően a titrálóedény (vezetőképességi cella) külsejére helyezik (felhelyezik), hogy ne érintkezzenek a titrálandó oldattal. A mérési eredmények alapján konduktometrikus titrálási görbe készül. A titrálás végpontját a titrálási görbe azon szakaszainak extrapolálásával találjuk meg, ahol a meredeksége változik. 4. FEJEZET KONDUKTOMETRIAI ELEMZÉS (CONDUCTOMETRY) 4.1 A módszer lényege A polarográfiai elemzés (polarográfia) egy külső potenciált alkalmazó elektrokémiai (jelen esetben polarográfiás) cella elektromos paraméterei és a benne lévő elemzett oldat tulajdonságai közötti összefüggések felhasználásán alapul. a) A kvalitatív polarográfiás analízis a mikroelektródán alkalmazott külső elektromos potenciál nagysága, amelynél az analit redukciója (vagy oxidációja) megfigyelhető adott körülmények között a mikroelektródán, és a redukálandó anyag természete (ill. oxidált). b) A kvantitatív polarográfiás analízis során a diffúziós elektromos áram nagysága és a vizsgált oldatban mért (redukáló vagy oxidáló) anyag koncentrációja közötti összefüggést használjuk. Az elektromos paraméterek - az alkalmazott elektromos potenciál nagysága és a diffúziós áram nagysága - a kapott polarizációs, vagyis áram-feszültség görbék elemzésével határozhatók meg, amelyek grafikusan tükrözik a polarográfiai cellában áramló elektromos áram függését a polarizációs cellában a mikroelektróda alkalmazott potenciálja. Ezért a polarográfiát néha direkt voltammetriának is nevezik. A higanycsepegő elektródával végzett klasszikus polarográfiás elemzési módszert 1922-ben Jaroslav Heyrovsky (1890-1967) cseh tudós fejlesztette ki és javasolta, bár magát a higanycsepegő elektródát B. Kucera cseh fizikus használta már 1903-ban. 1925-ben J. Heyrovsky és M. Shikata megtervezte az első polarográfot, amely lehetővé tette a polarizációs görbék automatikus rögzítését. Ezt követően a polarográfiás módszer különféle módosításait fejlesztették ki. Az átlagos diffúziós áram iD értékét a (25) Ilkovich-egyenlet határozza meg: ahol K az arányossági együttható, c a polarográfiailag aktív depolarizáló anyag koncentrációja (mmol/l); Az iD-t mikroamperben mérik a határáram és a maradékáram különbségeként. Az Ilkovich-egyenletben szereplő K arányossági együttható számos paramétertől függ, és egyenlő K=607nD12m23τ16 ahol n az elektród-redox reakcióban részt vevő elektronok száma; D a redukáló anyag diffúziós együtthatója (cm2/s); t a kapillárisból másodpercenként kiáramló higany tömege (mg); t egy higanycsepp képződési ideje (másodpercben) félhullámú potenciál mellett (általában 3-5 s). Mivel a D diffúziós együttható a hőmérséklettől függ, az Ilkovich-egyenletben szereplő K arányossági együttható a hőmérséklettel változik. A 20-50 °C hőmérséklet-tartományban lévő vizes oldatok esetében a polarográfiailag aktív depolarizáló anyagok diffúziós együtthatója körülbelül 3%-kal növekszik a hőmérséklet egy fokkal történő emelkedésével, ami az átlagos diffúziós áram iD ~1-gyel növekedéséhez vezet. -2%. Ezért a polarográfiát állandó hőmérsékleten végzik, a polarográfiai cellát általában 25 ± 0,5 ° C-on termosztálják. A higany tömege t és a cseppképződés ideje t függ a higanycsepp-elektróda jellemzőitől, valamint a kapillárisban és a kapillárishoz kapcsolódó tartályban lévő higanyoszlop magasságától. A higanycsepegő mikroelektród üvegkapillárisának külső átmérője általában 3-7 mm, belső átmérője 0,03-0,05 mm, hossza 6-15 cm A higanyoszlop magassága a kapilláris alsó végétől a tározó higanyfelületének felső szintjéig 40-80 cm; A vizsgált polarográfiás oldat indifferens elektrolit-tartalmának körülbelül 100-szor nagyobbnak kell lennie, mint a meghatározandó depolarizáló anyag tartalma, és a háttérelektrolit ionjai polarográfiás körülmények között nem ürülhetnek ki mindaddig, amíg a polarográfiailag aktív anyag ki nem ürül. A polarográfiát víz, víz-szerves keverékek (víz - etanol, víz - aceton, víz - dimetil-formamid stb.) és nemvizes közeg (etanol, aceton, dimetil-formamid, dimetil-szulfoxid stb.) oldószerrel végezzük. A polarográfia megkezdése előtt inert gázáramot (nitrogén, argon stb.) vezetnek át a vizsgált oldaton az oldott oxigén eltávolítására, amely szintén polarográfiai hullámot kelt a redukció miatt az alábbi séma szerint: 2Н+ + 2е = Н202 Н202 + 2Н+ + 2е = 2Н20 Időnként - lúgos oldatok esetén - inert gázáram átvezetése helyett a vizsgált oldathoz csekély mennyiségű aktív redukálószert - nátrium-szulfitot, metolt - adnak, amelyek azzal reagálva megkötik az oldott oxigént. 4.2 Kvantitatív polarográfiai elemzés A fentiekből következik, hogy a kvantitatív polarográfiás analízis a diffúziós áram iD mérésén alapul, a polarográfiailag aktív depolarizáló anyag polarográfiás oldatban meghatározott koncentrációjának függvényében. A kapott polarogramok elemzésekor az analit koncentrációját a kalibrációs görbe, a standard hozzáadások és a standard oldatok módszereivel határozzuk meg. a) Leggyakrabban a kalibrációs görbe módszert alkalmazzák. Ezzel a módszerrel standard oldatok sorozatát készítik, amelyek mindegyike pontosan ismert koncentrációjú analitot tartalmaz. Mindegyik oldatot polarografáljuk (miután inert gáz áramot fújunk át rajta) azonos körülmények között, polarogramokat kapunk, és megtaláljuk az E12 (minden oldatnál azonos) és az iD diffúziós áram értékeit (minden oldatnál eltérő) . A kapott adatok alapján iD-c koordinátákban kalibrációs gráfot készítünk, amely általában az Ilkovich-egyenletnek megfelelő egyenes. Ezután a vizsgált oldaton polarográfiát végzünk az ismeretlen c(X) analitkoncentrációval, polarogramot készítünk, megmérjük az iD(X) diffúziós áramot, és a kalibrációs grafikonon meghatározzuk a c(X) koncentrációt. . b) Standard összeadási módszer. A vizsgált oldat c(X) ismeretlen koncentrációjú analit polarogramját kapjuk, és meghatározzuk a diffúziós áram értékét, azaz. a polarogram h magassága. Ezután pontosan ismert mennyiségű analitot adunk a vizsgált oldathoz, növelve annak koncentrációját c(st) érték esetén ismét polarográfiát végeznek, és a diffúziós áram új értékét találjuk - a h + polarogram magasságát △ h. A (25) Ilkovich-egyenletnek megfelelően felírhatjuk: h = Kc(X), △ h = Kc(st), ahol h △ h = c(X)c(st) és c(X) = h △ hc(st) c) Standard megoldási módszer. Ugyanilyen körülmények között két oldatot polarografálunk: egy ismeretlen c(X) koncentrációjú vizsgálati oldatot és egy pontosan ismert c(st) koncentrációjú standard oldatot a meghatározandó anyagból. A kapott polarogramokból megtaláljuk a h(X) és h(st) polarográfiai hullámok magasságát, amelyek megfelelnek a c(X) és c(st) koncentrációjú diffúziós áramnak. Az Ilkovich-egyenlet (25) szerint: (X) = Kc(X), h(st) = Kc(st), A standard oldatot úgy készítjük el, hogy koncentrációja a lehető legközelebb legyen a meghatározandó oldat koncentrációjához. Ilyen körülmények között a meghatározási hiba minimális. 3 A polarográfia alkalmazásai A módszer alkalmazása. A polarográfiát kis mennyiségű szervetlen és szerves anyag meghatározására használják. Több ezer kvantitatív polarográfiai elemzési technikát fejlesztettek ki. Szinte az összes fémkation, számos anion (bróm-, jodát-, nitrát-, permanganát-ion), diazo-csoportokat tartalmazó szerves vegyületek, karbonil-, peroxid-, epoxicsoportokat, kettős szén-szén kötések polarográfiai meghatározására javasoltak módszereket. , valamint szén-halogén, nitrogén-oxigén, kén-kén köt. A gyógyszerkönyvi módszerrel szalicilsav, norszulfazol, B-vitamin alkaloidok, folsav, kellin porban és tablettában, nikotinamid, piridoxin-hidroklorid, arzénkészítmények, szívglikozidok, valamint a gyógyszerekben található oxigén és különféle szennyeződések határozhatók meg. A módszer nagy érzékenységgel rendelkezik (10"5-10T6 mol/l-ig); szelektivitása; az eredmények viszonylag jó reprodukálhatósága (akár ~2%); széles körű alkalmazási körrel rendelkezik; lehetővé teszi az anyagok keverékeinek elemzését azok szétválasztása nélkül, színezett oldatok, kis térfogatú oldatok (a polarográfiai sejtek térfogata akár 1 ml is lehet); az elemzést oldatáramban végezze; automatizálja az elemzést." A módszer hátrányai közé tartozik a higany toxicitása, meglehetősen könnyű oxidációja oxidáló anyagok jelenlétében, valamint az alkalmazott berendezések viszonylagos bonyolultsága. A polarográfiás módszer egyéb változatai. A fent ismertetett klasszikus polarográfián kívül, amely egy csepegtető higany mikroelektródát használ, amelynek elektromos potenciálja állandó elektromos áram mellett egyenletesen növekszik, a polarográfiás módszer további változatait fejlesztették ki - derivált, differenciál-, impulzus-, oszcillográfiás polarográfia; váltóáramú polarográfia - különböző változatokban is. 5. FEJEZET Amperometrikus TITRÁLÁS A módszer lényege. Az amperometrikus titrálás (potenciosztatikus polarizációs titrálás) a voltammetriás módszer egyik fajtája (a polarográfiával együtt). Egy elektrokémiai cella elektródái közötti áramerősség mérésén alapul, amelyre bizonyos feszültséget kapcsolnak, a hozzáadott titrálószer térfogatának függvényében. Az Ilkovich-egyenlet (25) szerint: Minél nagyobb a polarográfiailag aktív anyag c koncentrációja, annál nagyobb a diffúziós áram iD a polarográfiai cellában. Ha a polarográfiás cellában elhelyezett elemzett titrált oldathoz titrálót adva csökken vagy nő az ilyen anyag koncentrációja, akkor a diffúziós áram is ennek megfelelően csökken vagy nő. Az ekvivalencia pontot a diffúziós áram csökkenésének vagy növekedésének éles változása határozza meg, amely megfelel a titrált anyag és a titrálószer reakciójának végének. Megkülönböztetik az egy polarizálható elektródával végzett amperometrikus titrálást, amelyet áramkorlátozó titrálásnak, polarográfiai vagy polarimetriás titrálásnak is neveznek, és két azonos polarizálható elektróddal végzett amperometrikus titrálást, vagy „amíg az áram teljesen meg nem áll”, biamperometrikus titrálást. Amperometrikus titrálás egy polarizálható elektródával. Ez egy polarográfiai cellában az áram mérésén alapul, a hozzáadott titrálószer mennyiségétől függően a mikroelektródon állandó külső potenciál mellett, valamivel nagyobb, mint a titrált X anyag vagy T titráló áram-feszültség görbéjén lévő félhullám potenciál. Jellemzően a kiválasztott külső potenciál az X vagy T polarogramon lévő határáram tartományának felel meg. A titrálást egy állítható feszültségű egyenáramú forrásból álló berendezésen végzik, amelyhez galvanométer és polarográfiai titráló cella van sorba kötve. . A cella munka (jelző) elektródája lehet higanycsepp elektróda, álló vagy forgó platina vagy grafit elektróda. Szilárd elektródák használata esetén a titrálás során az oldatot keverni kell. Referenciaelektródaként ezüst-klorid vagy kalomel elektródákat használnak. A háttérben a körülményektől függően különböző polarográfiailag inaktív elektrolitok állnak adott potenciálon (HN03, H2S04, NH4NO3 stb.). Először áram-feszültség görbéket (polarogramokat) kapunk X-re és T-re ugyanazon feltételek mellett, amelyek mellett az amperometrikus titrálást el kell végezni. Ezen görbék figyelembevételével kiválasztunk egy potenciálértéket, amelynél a polarográfiailag aktív X vagy T határáramértékét elérjük. Az amperometrikus titráláshoz használt T titráló koncentrációnak körülbelül 10-szer nagyobbnak kell lennie, mint az X koncentráció; ebben az esetben gyakorlatilag nincs szükség az oldat hígítására vonatkozó korrekció bevezetésére a titrálás során. Ellenkező esetben a polarogramok készítéséhez szükséges összes feltétel teljesül. A termosztáttal szemben támasztott követelmények kevésbé szigorúak, mint a közvetlen polarográfiánál, mivel a titrálás végét nem a diffúziós áram abszolút értéke határozza meg, hanem annak éles változása. Az X-et tartalmazó elemzett oldatot a polarográfiás cellába, és a T titrálószert kis részletekben, minden alkalommal megmérve az i áramot. Az i áram nagysága a polarográfiailag aktív anyag koncentrációjától függ. Az ekvivalencia ponton az i értéke élesen változik. Az amperometrikus titrálás eredményei alapján titrálási görbék készülnek. Az amperometrikus titrálási görbe az áram változásának grafikus ábrázolása / a hozzáadott titráló V térfogatának függvényében. A titrálási görbét az áram i - a hozzáadott T titráló V térfogata (vagy a titrálás mértéke) koordinátáiban ábrázoljuk. A titrált X anyag és a T titrálószer természetétől függően az amperometrikus titrálási görbék különböző típusúak lehetnek. A biamperometrikus titrálást az oldat erőteljes keverésével, egy potenciométeres egyenáramú forrásból álló elrendezésben végezzük, amelyből érzékeny mikroamperméteren keresztül állítható potenciálkülönbséget (0,05-0,25 V) juttatunk az elektrokémiai cella elektródáira. A titrálás előtt a titrálandó oldatot ez utóbbihoz adjuk, és a titrálót részletekben adagoljuk, amíg az áram hirtelen meg nem áll vagy meg nem jelenik, a mikroampermérő leolvasása alapján. Az elektrokémiai cellában használt platina elektródákat időszakonként tisztítjuk, ~30 percre forrásban lévő, vas-klorid adalékokat tartalmazó tömény salétromsavba merítjük, majd az elektródákat vízzel mossuk. A biamperometrikus titrálás gyógyszerkönyvi módszer; jodometriában, nitritometriában, akvametriában és nem vizes közegben történő titráláshoz használják. 6. FEJEZET KOULOMETRIAI ELEMZÉS (COULOMETRIA) 1 A módszer alapelvei elektrokémiai konduktometria titrálás coulometria A coulometriás elemzés (coulometria) az elektrokémiai cellában elektrolízis során reakcióba lépő anyag m tömege és az elektrokémiai cellán csak ennek az anyagnak az elektrolízise során áthaladó Q elektromosság mennyisége közötti összefüggésen alapul. Faraday egységes elektrolízistörvényének megfelelően az m tömeget (grammban) a (27) összefüggés alapján a Q elektromosság mennyiségével (coulombban) viszonyítjuk. ahol M az elektrolízis során reakcióba lépő anyag moláris tömege, g/mol; n az elektródreakcióban részt vevő elektronok száma; 96487 C/mol a Faraday-szám. Az elektrolízis során egy elektrokémiai cellán áthaladó Q elektromosság (C-ben) megegyezik az i elektromos áram (A-ban) és az elektrolízis idejének szorzatával. τ ( c): Ha megmérjük a Q villamos energia mennyiségét, akkor a (27) szerint számítható az m tömeg, ez arra az esetre igaz, ha az elektrolízis során az elektrokémiai cellán áthaladó Q villamos energia teljes mennyiségét csak egy elektrolízisre fordítjuk. adott anyag; mellékfolyamatokat ki kell zárni. Más szavakkal, az áramteljesítménynek (hatékonyságnak) 100%-nak kell lennie. Mivel M. Faraday egységes elektrolízistörvényének megfelelően az elektrolízis során reagált anyag tömegének t (g) meghatározásához meg kell mérni a meghatározandó anyag elektrokémiai átalakítására fordított Q elektromosság mennyiségét. , coulombban, a módszert coulometriának nevezik. A coulometriás mérések fő feladata a Q elektromosság mennyiségének minél pontosabb meghatározása. A coulometriás analízist vagy amperosztatikus (galvanosztatikus) módban, pl. állandó elektromos árammal i=const, vagy a munkaelektróda szabályozott állandó potenciáljával (potenciosztatikus coulometria), amikor az elektromos áram az elektrolízis folyamata során változik (csökken). Az első esetben a Q elektromosság mennyiségének meghatározásához elegendő a t(s) elektrolízisidőt, az egyenáramot /(A) a lehető legpontosabban megmérni, és a (10.28) képlet segítségével kiszámítani a Q értékét. A második esetben a Q értékét számítással vagy kémiai coulométerrel határozzuk meg. Létezik direkt coulometria és indirekt coulometria (kulometriás titrálás). A módszer lényege. A közvetlen coulometriát állandó áram mellett ritkán alkalmazzák. Gyakrabban a kulometriát a munkaelektróda szabályozott állandó potenciáljával vagy közvetlen potenciosztatikus coulometriával alkalmazzák. Közvetlen potenciosztatikus coulometriában a meghatározandó anyagot közvetlenül elektrolizálják. Megmérik az anyag elektrolízisére fordított villamos energia mennyiségét, és az egyenlet segítségével kiszámítják a meghatározandó anyag m tömegét. Az elektrolízis folyamata során a munkaelektróda potenciálját állandó értéken tartják, E = const, amelyhez általában eszközöket - potenciosztátokat - használnak. Az E potenciál konstans értékét előre kiválasztják az áram-feszültség (polarizációs) görbe figyelembevételével, amelyet az áram i - E potenciál koordinátákban konstruálnak (ahogyan a polarográfiában történik), amelyet ugyanolyan körülmények között kapunk, mint az elektrolízis. végrehajtani. Általában egy olyan E potenciálértéket választanak ki, amely megfelel az elemzett anyag határáram-tartományának, és valamivel magasabb, mint az E12 félhullámpotenciál (-0,05-0,2 V-tal). Ennél a potenciálértéknél, mint a polarográfiánál, a háttér-elektrolit nem mehet elektrolízisen. Ahogy az elektrolízis folyamata állandó potenciálon megy végbe, az elektromos áram a cellában csökken, ahogy az elektródreakcióban részt vevő elektroaktív anyag koncentrációja csökken. Ebben az esetben az elektromos áram idővel egy exponenciális törvény szerint csökken a kezdeti i0 értékről a t = O időpontban a t időpontban lévő i értékre: ahol a k együttható a reakció természetétől, az elektrokémiai cella geometriájától, a munkaelektróda területétől, a meghatározandó anyag diffúziós együtthatójától, az oldat keverési sebességétől és térfogatától függ. Az oldaton áthaladó elektromosság mennyiségének meghatározására szolgáló módszerek direkt potenciosztatikus kulometriában. A Q értéke számítási módszerekkel vagy kémiai kulométerrel határozható meg. a) A Q mennyiség kiszámítása az i versus m görbe alatti területből A Q észrevehető hiba nélküli meghatározásához a módszer az elektrolízis folyamatának majdnem teljes befejezését igényli, azaz. hosszú ideje. A gyakorlatban, amint fentebb megjegyeztük, a területet m értékkel mérik 0,001i0 (i0 0,1%-a). b) Q értékének kiszámítása In / m-től való függése alapján. Ennek megfelelően a következőket kapjuk: Q = 0∞i0e-k τ d τ =i00∞e-k τ d τ =i0k Mert a ∞i0e-k τ d τ = - k-1 e-k∞-e-k0= k-10-1=k-1 A direkt coulometria alkalmazása. A módszer nagy szelektivitással, érzékenységgel (10-8-10-9 g-ig vagy ~10-5 mol/l-ig), reprodukálhatóságával (~1-2%-ig) rendelkezik, és lehetővé teszi a mikroszennyeződés-tartalom meghatározását. A módszer hátrányai közé tartozik az elemzés nagy munkaintenzitása és időtartama, valamint a drága berendezések szükségessége. Közvetlen kulometriával meghatározható - katódos redukció során - fémionok, szerves nitro- és halogénszármazékok; anódos oxidáció során - klorid, bromid, jodid, tiocianát anionok, fémionok alacsonyabb oxidációs állapotban, amikor magasabb oxidációs állapotba alakítják át, pl.: As(IH) -> As(V),Cr(II) - > Cr(III) ), Fe(II) -» Fe(III), T1(I) -> Tl(III) stb. A gyógyszerészeti elemzésben direkt coulometriát alkalmaznak az aszkorbinsav és a pikrinsav, a novokain, a hidroxi-kinolin és néhány más esetben meghatározására. Amint fentebb megjegyeztük, a közvetlen coulometria meglehetősen munka- és időigényes. Ezenkívül bizonyos esetekben a mellékfolyamatok már a fő elektrokémiai reakció befejeződése előtt észrevehetően megjelennek, ami csökkenti az áram hatékonyságát és jelentős elemzési hibákhoz vezethet. Ezért gyakrabban alkalmazzák az indirekt coulometriát - coulometriás titrálást. 3 Kulonometrikus titrálás A módszer lényege. A coulometriás titrálás során az elektrokémiai cellában oldatban lévő X analit reakcióba lép a T „titrálóval”, az oldatban szintén jelen lévő segédanyag elektrolízise során a generátorelektródán folyamatosan képződő (generálódó) anyaggal. A titrálás vége - az a pillanat, amikor az összes X analit teljesen reagál a keletkezett „titráló” T-vel, vagy vizuálisan rögzítjük indikátor módszerrel, megfelelő indikátort juttatva az oldatba, amely megváltoztatja a színét a TE közelében, vagy műszeres módszerekkel. - potenciometrikusan, amperometrikusan, fotometriailag. Így a coulometriás titrálásnál a titrálószert nem adják a bürettából a titrálandó oldathoz. A titráló szerepét a T anyag tölti be, amely az elektróda reakciója során folyamatosan keletkezik a generátor elektródán. Nyilvánvalóan van analógia a hagyományos titrálás között, amikor a titrálószert kívülről vezetik be a titrált oldatba, és amikor hozzáadják, reakcióba lép a vizsgálandó anyaggal, valamint a T anyag keletkezése között, amely képződésekor szintén reagál. az analittal. Ezért a vizsgált módszert „kulométeres titrálásnak” nevezik. A coulometriás titrálást amperosztatikus (galvanosztatikus) vagy potenciosztatikus üzemmódban végezzük. A coulometriás titrálást gyakrabban amperosztatikus üzemmódban végzik, miközben az elektromos áramot az elektrolízis teljes ideje alatt állandó szinten tartják. A kulométeres titrálásnál a hozzáadott titrálószer térfogata helyett a t időt és az elektrolízis áramát mérjük. A T anyag képződését a coulometriás cellában az elektrolízis során titrálóképzésnek nevezzük. Kulonometrikus titrálás állandó áram mellett. A coulometriás titrálás során amperosztatikus üzemmódban (állandó áramerősség mellett) megmérjük azt a t időt, amely alatt az elektrolízis végbement, és a képlet segítségével kiszámítjuk az elektrolízis során elfogyasztott Q elektromos áram mennyiségét, amely után meghatározzuk az X analit tömegét az arány. Így például a HC1 sósav oldatának kulometriás titrálási módszerrel történő standardizálását a HC1 tartalmú standardizált oldat H30+ hidrogénionjainak titrálásával hajtják végre, amelyet a platina katódon elektromosan generálnak az OH- hidroxidionok az elektrolízis során. víz: Н20 + 2е = 20Н- + Н2 A keletkező titrálószer - hidroxidionok - reakcióba lép az oldatban lévő H30+ ionokkal: H30+ + OH- = 2H20 A titrálást fenolftalein indikátor jelenlétében végezzük, és leállítjuk, amikor az oldat halvány rózsaszínű színe van. Ismerve az i egyenáram nagyságát (amperben) és a titrálásra fordított t időt (másodpercben), a Q elektromosság mennyiségét (coulombban) a (28) képlet segítségével számítjuk ki, és a reagált HC1 tömegét (grammban) tartalmazza. a (27) képletben) a kulometriás cellához (a generátor edénybe) hozzáadott standardizált HC1 oldat alikvot részében. A coulometriás titrálás feltételei. A fentiekből következik, hogy a coulometriás titrálás feltételeinek 100%-os áramhatékonyságot kell biztosítaniuk. Ehhez teljesítenie kell legalább a következő követelményeket. a) A segédreagensnek, amelyből a munkaelektródán a titrálót előállítják, a vizsgálandó anyaghoz képest nagy feleslegben kell jelen lennie az oldatban (~ 1000-szeres felesleg). Ilyen körülmények között általában megszűnnek az elektrokémiai mellékreakciók, amelyek közül a fő a háttérelektrolit, például a hidrogénionok oxidációja vagy redukciója: Н+ + 2е = Н2 b) Az elektrolízis során az i=const egyenáram értékének kisebbnek kell lennie, mint a segédreagens diffúziós áramának értéke, hogy elkerüljük a háttérelektrolit ionjaival járó reakciót. c) A lehető legpontosabban meg kell határozni az elektrolízis során elfogyasztott villamos energia mennyiségét, amihez az időszámlálás kezdetének és végének, valamint az elektrolízisáram nagyságának pontos rögzítése szükséges. Kulometriás titrálás állandó potenciálon. A potenciosztatikus módot ritkábban használják coulometriás titrálásnál. A potenciosztatikus üzemmódban végzett coulometriás titrálást egy anyag munkaelektródon lévő kisülési potenciáljának megfelelő állandó potenciálértéken hajtják végre, például az M "* fémkationok katódos redukciója során egy platina munkaelektródán. halad, a potenciál mindaddig állandó marad, amíg az összes fémkation reakcióba lép, majd hirtelen csökken, mivel az oldatban már nincsenek potenciált meghatározó fémkationok. Kulometriás titrálás alkalmazása. A coulometriás titrálásban minden típusú titrimetriás analízisreakció alkalmazható: sav-bázis, redox, precipitációs, komplexképző reakciók. Így kis mennyiségű savat lehet meghatározni coulometriás sav-bázis titrálással a víz katódon történő elektrolízise során keletkező elektrogenerált OH-ionokkal: Н20 + 2е = 20Н" + Н2 A bázisok a víz elektrolízise során az anódon keletkező H+ hidrogénionokkal is titrálhatók: Н20-4е = 4Н+ + 02 Redox bromometriás coulometriás titrálással arzén(III), antimon(III), jodidok, hidrazin, fenolok és egyéb szerves anyagok vegyületei határozhatók meg. A bróm, amely elektromosan keletkezik az anódon, titrálószerként működik: VG -2e = Vg2 A precipitatív coulometriás titrálással meghatározhatók a halogenidionok és szerves kéntartalmú vegyületek elektrogenerált ezüstkationokkal Ag+, cinkkationok Zn2+ elektrogenerált ferrocianidionokkal stb. A fémkationok komplexometriás coulometriás titrálása elvégezhető higany(I)-komplexonát katódon elektrogenerált EDTA-anionokkal. A coulometriás titrálás nagy pontossággal rendelkezik, a kvantitatív elemzésben széles körben alkalmazható, lehetővé teszi kis mennyiségű anyagok, alacsony rezisztenciájú vegyületek meghatározását (mivel képződésük után azonnal reagálnak), például réz (1), ezüst (H) ón (P), titán (III), mangán (III), klór, bróm stb. A módszer előnyei közé tartozik az is, hogy a titrálószer előkészítése, standardizálása és tárolása nem szükséges, mivel az elektrolízis során folyamatosan képződik, és a meghatározandó anyaggal való reakcióban azonnal elfogy. KÖVETKEZTETÉS Az elektrokémiai elemzési módszerek az elektródákon vagy az elektródák közötti térben végbemenő folyamatokon alapulnak. Az elektrokémiai elemzési módszerek a legrégebbi fizikai-kémiai elemzési módszerek közé tartoznak (néhányat a 19. század végén írtak le). Előnyük a berendezések és az elemzési technikák nagy pontossága és viszonylagos egyszerűsége. A nagy pontosságot az elektrokémiai elemzési módszerekben használt nagyon pontos törvények határozzák meg, például a Faraday-törvény. Nagy kényelem, hogy elektromos hatásokat alkalmaznak, és az a tény, hogy ennek a hatásnak az eredményét (választ) kapják meg elektromos jel formájában. Ez biztosítja az olvasás nagy sebességét és pontosságát, és széles körű automatizálási lehetőségeket nyit meg. Az elektrokémiai elemzési módszereket jó szenzitivitás és szelektivitás jellemzi, esetenként a mikroanalízishez sorolhatók, mivel esetenként 1 ml-nél kevesebb oldat is elegendő az elemzéshez. Műszerük egy elektrokémiai cella, amely egy elektrolitoldattal ellátott edény, amelybe legalább két elektródát merítenek. A megoldandó problémától függően az edény alakja és anyaga, az elektródák száma és jellege, az oldat, valamint az elemzés körülményei (a rákapcsolt feszültség (áram) és a rögzített analitikai jel, hőmérséklet, keverés, inert gázzal történő átöblítés stb.). ) változhat. A meghatározandó anyag része lehet mind a cellát kitöltő elektrolitnak, mind az egyik elektródának. Az elektrokémiai elemzési módszerek fontos szerepet játszanak a modern világban. Korunkban különösen fontos a környezetvédelem. Ezekkel a módszerekkel nagyszámú különböző szerves és szervetlen anyag tartalmát lehet meghatározni. Ma már hatékonyabban azonosítják a veszélyes anyagokat.
Küldje el a jó munkát a tudásbázis egyszerű. Használja az alábbi űrlapot
Diákok, végzős hallgatók, fiatal tudósok, akik a tudásbázist tanulmányaikban és munkájukban használják, nagyon hálásak lesznek Önnek.
közzétett http://www.allbest.ru
Az Orosz Föderáció Oktatási és Tudományos Minisztériuma
Szövetségi Állami Költségvetési Oktatási Intézmény
felsőoktatás
"Irkutszki Nemzeti Kutató Műszaki Egyetem"
Színesfémkohászati Tanszék
(osztály neve)
"Elektrokémiai kutatási módszerek"
Absztrakt a tudományágról
„Fizikai-kémiai módszerek a kohászati folyamatok tanulmányozására”
Az MCM-16-1 csoport diákja fejezte be
Zakharenkov R.I.
Az MCM tanszék tanára ellenőrizte
Kuzmina M.Yu.
Irkutszk 2017
BEVEZETÉS
Az elektrokémia a fizikai kémia olyan ága, amely az ionokat tartalmazó rendszereket (elektrolitok oldatait vagy olvadékait) és a két fázis határán, töltött részecskék részvételével zajló folyamatokat vizsgálja.
Az első elképzelések a kémiai és elektromos jelenségek kapcsolatáról a 18. században ismertek, hiszen rengeteg fizikai és kémiai kísérletet végeztek elektromos és villámkisülésekkel, töltésekkel Leyden tégelyekben, de ezek mindegyike véletlenszerű volt. természet az állandó erős elektromos energiaforrás hiánya miatt. Az elektrokémia eredete L. Galvani és A. Volta nevéhez fűződik. A béka élettani funkcióinak tanulmányozása közben Galvani véletlenül elektrokémiai áramkört hozott létre. Két különböző fémből és egy előkészített békacombból állt. A mancs elektrolit és elektromos áram mutatója is volt, de a következtetést hibásan adták meg, vagyis Galvani szerint ez az áramkörben keletkezett elektromos áram állati eredetű volt, vagyis az áramkör funkcionális jellemzőivel függött össze. béka teste ("állati elektromosság" elmélet).
Galvani kísérleteinek helyes értelmezését A. Volta adta meg. Megalkotta az első galvánelem akkumulátort - egy voltikus oszlopot. Az akkumulátorcellák réz- és cinktárcsákból álltak, az elektrolit pedig sós vízzel vagy savval átitatott szivacsanyag volt. Ez a kapcsolat tette lehetővé az elektromos áram elérését. A nagy tudósok, A. Volta, J. Daniel, B. S. Jacobi, P. R. Bagration, G. Plante és mások munkái révén hamarosan megjelentek a nagy teljesítményű, könnyen használható galváncellák és akkumulátorok. Aztán A. Volta kifejlesztett egy sorozat fémfeszültséget. Ha két különböző fémet érintkezésbe hozunk, majd szétválasztunk, akkor fizikai eszközökkel, például elektroszkóppal látható, hogy az egyik fém pozitív, a másik negatív töltést kapott. Ez a fémsorozat, amelyben minden előző fém pozitív töltésű, de bármely következővel, azaz a Volta sorozattal való érintkezés után hasonlónak bizonyult a feszültségsorokhoz.
Továbbá, a 19. század elején kifejlesztették az elektrolízist, és M. Faraday megállapította az elektrolízis mennyiségi törvényeit. A tudósok nagymértékben hozzájárultak az elektrokémia fejlődéséhez: S. A. Arrhenius, V. F. Ostwald, R. A. Colley, P. Debye, W. Nernst, G. Helmholtz stb. Az elektrokémia jelenleg elméleti és alkalmazott részekre oszlik. Az elektrokémiai módszerek alkalmazásával összekapcsolódik a fizikai kémia más ágaival, valamint az analitikus kémiával és más tudományokkal.
elektrokémiai potenciometria konduktometria coulometria
1 . ELEKTROKÉMIAI KUTATÁSI MÓDSZEREK
Az elektrokémiai folyamatok tanulmányozására különféle módszerek alkalmazásának szükségessége az elektródreakciókban az elektrontranszfer sebességének széles skálájának köszönhető. Mindegyik módszernek van egy bizonyos határa a csereáram-sűrűség meghatározott értékére, amely felett az elektródreakció elektrokémiai paraméterei nem határozhatók meg. Minden egyes objektum kapcsán ki kell választani azt a módszert, amely a lehető legtöbb megbízható információt nyújtja. Az elektrokémiai vizsgálatok elvégzésekor ismerni kell a kiindulási anyagok és reakciótermékek kémiai összetételét. Az elektrolit összetételének meghatározásához különféle fizikai és kémiai módszereket alkalmaznak: spektrofotometriás, potenciometrikus, analitikai és mások. Az elektrokémiai vizsgálatok elvégzésekor a következő feltételeket kell betartani.
1. A felhasznált reagensek maximális tisztasága; szigorúan ismerni kell az elektródák összetételét, valamint felületeik állapotát. Ügyelni kell arra, hogy az elektródák felülete ne változzon a mérési folyamat során.
2. Az elektrokémiai cella és a benne elhelyezett elektródák kialakításának biztosítania kell az áram egyenletes eloszlását a munkaelektróda teljes felületén.
3. A méréseket szigorúan ellenőrzött hőmérsékleten kell elvégezni.
4. Tartsa állandó nyomást és a gázfázis összetételét az elektrolit felett. A vizsgálatokat általában inert gáz környezetben (N 2, Ar, Ne, He H 2) végezzük, mivel a gázfázisban lévő oxigén jelentős hatással lehet a folyamat mechanizmusára.
5. Olyan kísérleti körülményeket kell biztosítani, amelyek mellett az elektromos kettős réteg diffúz részében a potenciálesés minimális vagy pontosan ismert lenne. Ennek a potenciálnak a csökkentésére rendszerint háttérelektrolitot használnak, amelynek koncentrációja legalább 20-szor nagyobb, mint a fő anyagé. Először azonban meg kell győződnie arról, hogy a háttér elektrolit nem torzítja a vizsgált reakció polarizációs görbéjét.
6. A munkaelektróda potenciáljának pontos mérése. Ehhez meg kell szüntetni a diffúziós potenciált a vizsgált elektrolit és a referenciaelektród elektrolitja között. Ez a potenciál a határáramhoz közeledve veszi fel a maximális értékét, és jelentősen torzíthatja a mérési eredményeket. A vizsgált elektrolit és a referenciaelektród elektrolitja közötti diffúziós potenciál kiküszöbölése érdekében kívánatos: a) olyan referenciaelektródot válasszunk, amelynek elektrolit-összetétele megegyezik a vizsgált elektrolit-összetétellel. Például a kloridos oldatok kutatása során célszerű ezüst-klorid-, kalomel- és klórelektródákat használni; savas szulfát oldatokban - higany-szulfát elektródák stb.; b) használjon referenciaelektródát olyan elektrolittal, amelynek határán a vizsgált elektrolittal ismert képletekkel kiszámítható a diffúziós potenciál.
Állandó ionerősségű oldatokban és magas háttérkoncentrációknál - állandó ionkoncentráció mellett - elvileg bármilyen referenciaelektródát használhat. A diffúziós potenciál ebben az esetben lehet nagyon nagy, de állandó is - kiszámítható vagy kísérletileg meghatározható.
Az elektrokémiai folyamatok kinetikájának vizsgálatakor minden esetben szükséges az áramsűrűség mérése. Általában analitikai kémiai és coulometriás módszerekkel kezdik annak megállapítását, hogy csak egy vizsgált reakció játszódik le az elektródán, vagy azt mellékreakciók bonyolítják-e. Mellékreakciók esetén azt kell kideríteni, hogy az áram mekkora hányada származik csak a vizsgált reakció megvalósulásából (konstruálja meg a vizsgált reakcióra az ún. parciális polarizációs karakterisztikát).
Az elektródreakció mechanizmusa a legegyszerűbben csak abban az esetben értelmezhető, ha a kiindulási anyagot 100%-os áramhatékonysággal egy termékké alakítják. A reakció Faraday törvényének való megfelelésének ellenőrzése vagy a coulometriás mérések elvégzése lehetővé teszi, hogy egyidejűleg meghatározza a teljes elektródreakcióban részt vevő elektronok számát. A kiindulási anyag és a reakciótermék összetételének, valamint az átvitt elektronok teljes számának ismerete lehetővé teszi a teljes elektródreakció egyenletének felírását.
Az elektródreakció mechanizmusának tanulmányozásának következő lépése annak megállapítása, hogy melyik szakasz korlátozza.
Ha a korlátozó szakasz a kisülési-ionizációs szakasz, és az összes többi reverzibilisen megy végbe, akkor a folyamat fő kinetikai paraméterei grafikusan vagy analitikusan meghatározhatók a lassú kisülés elméletének egyenleteinek a polarizációs jellemzőkre történő alkalmazásával.
1.1 Elektrokémiai elemzési módszerek
Elektrokémiai módszerekelemzés a kvalitatív és kvantitatív elemzési módszerek összessége, amelyek a vizsgált közegben vagy a határfelületen előforduló elektrokémiai jelenségeken alapulnak, és az elemzett anyag szerkezetének, kémiai összetételének vagy koncentrációjának változásaihoz kapcsolódnak.
Vannak direkt és közvetett elektrokémiai módszerek. A közvetlen módszerek az áramerősség (potenciál stb.) függését használják a meghatározandó komponens koncentrációjától. Az indirekt módszereknél az áramerősséget (potenciált stb.) mérik annak érdekében, hogy megtalálják az analit titrálásának végpontját megfelelő titrálóval, pl. A mért paraméternek a titráló térfogattól való függését használjuk.
Bármilyen elektrokémiai méréshez elektrokémiai áramkörre vagy elektrokémiai cellára van szükség, melynek szerves részét képezi a vizsgált oldat.
Az elektrokémiai módszereket az elemzési folyamat során mért jelenségek típusától függően osztályozzák. Az elektrokémiai módszereknek két csoportja van:
1. Módszerek külső potenciál bevezetése nélkül, a potenciálkülönbség mérésén alapulnak, amely egy elektródából és a tesztoldatot tartalmazó edényből álló elektrokémiai cellában jelentkezik. Ezt a módszercsoportot ún potenciometrikus. A potenciometriai módszerek az elektródák egyensúlyi potenciáljának az elektródákon zajló elektrokémiai reakcióban résztvevő ionok koncentrációjától való függését használják fel.
2. Mérésen alapuló, külső potenciál kivetésével járó módszerek:
a) Oldatok elektromos vezetőképessége? konduktometria;
b) Az oldaton áthaladó elektromosság mennyisége? coulometria;
c) Az áramerősség függése az alkalmazott potenciáltól? volt-amperometria;
d) Az elektrokémiai reakció lezajlásához szükséges idő - kronoelektrokémiai módszerek(chronovoltammemetria, kronokonduktometria).
Ennek a csoportnak a módszereiben az elektrokémiai cella elektródáira külső potenciált alkalmaznak.
Az elektrokémiai analízis műszereinek fő eleme az elektrokémiai cella. Olyan módszerekben, amelyek nem kényszerítenek ki idegen potenciált galvánelem, amelyben kémiai redox reakciók következtében elektromos áram keletkezik. Egy cellában, például galvanikus cellában, két elektróda érintkezik a vizsgált oldattal - egy indikátorelektróda, amelynek potenciálja az anyag koncentrációjától függ, és egy állandó potenciálú elektróda - egy referenciaelektróda, amellyel szemben a mérjük az indikátorelektróda potenciálját. A potenciálkülönbséget speciális eszközökkel - potenciométerekkel - mérik.
Az idegen potenciált kikényszerítő módszereknél, elektrokémiai cella, így nevezték el, mert a cella elektródáin egy alkalmazott potenciál hatására elektrolízis megy végbe - egy anyag oxidációja vagy redukciója. A konduktometriai elemzés során konduktometrikus cellát használnak, amelyben az oldat elektromos vezetőképességét mérik. Az elektrokémiai módszerek az alkalmazási mód szerint direkt módszerekre sorolhatók, amelyeknél az anyagok koncentrációját a készülék leolvasása alapján mérik, illetve elektrokémiai titrálásra, ahol elektrokémiai mérésekkel rögzítik az ekvivalenciapont jelzését. Ennek az osztályozásnak megfelelően megkülönböztetik a potenciometriát és a potenciometriás titrálást, a konduktometriát és a konduktometrikus titrálást stb.
Az elektrokémiai meghatározások eszközei az elektrokémiai cellán, a keverőn és a terhelési ellenálláson kívül tartalmaznak potenciálkülönbséget, áramerősséget, oldatellenállást és elektromos áram mennyiségét mérő eszközöket. Ezeket a méréseket mutató műszerekkel (voltmérő vagy mikroampermérő), oszcilloszkópokkal és automatikus rögzítő potenciométerekkel lehet elvégezni. Ha a cellából érkező elektromos jel nagyon gyenge, akkor azt rádióerősítők segítségével erősítik. Az idegen potenciált kiváltó módszerek eszközeiben fontos szerepet játszik a cella megfelelő potenciállal, stabilizált egyen- vagy váltakozó árammal való ellátása (a módszer típusától függően). Az elektrokémiai elemző készülékek tápegysége általában egy egyenirányítót és egy feszültségstabilizátort tartalmaz, amely biztosítja a készülék állandó működését.
1.2 Potenciometria
A potenciometria a meghatározandó anyagot tartalmazó oldatba merített eltérő elektródák közötti elektromos potenciálok különbségének mérésén alapul. Az elektródákon elektromos potenciál keletkezik, amikor redox (elektrokémiai) reakció megy végbe rajtuk. Redox reakciók mennek végbe egy oxidálószer és egy redukálószer között redox párok képződésével, amelyek E potenciálját a Nernst-egyenlet határozza meg az [ok] és [rec] párkomponensek koncentrációjával:
Ahol E°- standard elektródpotenciál, V;
n- a folyamatban részt vevő elektronok száma.
A potenciometrikus méréseket úgy végezzük, hogy két elektródát engedünk az oldatba - egy indikátorelektródát, amely reagál a meghatározandó ionok koncentrációjára, és egy standard vagy referenciaelektródát, amelyhez képest az indikátor potenciálját mérik. Többféle indikátor- és szabványelektródát használnak.
Az első típusú elektródák reverzibilis az elektródát alkotó fémionokhoz képest. Ha egy ilyen elektródát fémkationokat tartalmazó oldatba engedünk, egy elektródapár képződik: M n + /M.
Második típusú elektródák érzékenyek az anionokra, és az M fémet anionos oldhatatlan sójának rétegével vonják be A- amelyre az elektróda érzékeny. Amikor egy ilyen elektród érintkezik a megadott aniont tartalmazó oldattal A-, E potenciál keletkezik, melynek értéke a sóoldékonyság szorzatától függ
STB M.A. és anionkoncentráció [ A-] megoldásban.
A második típusú elektródák ezüst-klorid és kalomel. A telített ezüst-klorid és kalomel elektródák állandó potenciált tartanak fenn, és referenciaelektródákként használják őket, amelyekhez képest mérik az indikátorelektród potenciálját.
Inert elektródák- nehezen oxidálható fémekből - platina, arany, palládium - készült lemez vagy huzal. E mérésére szolgálnak redoxpárt tartalmazó oldatokban (például Fe 3+ /Fe 2+).
Membrán elektródák Különböző típusoknak van egy membránja, amelyen fellép a membránpotenciál E. Az E értéke attól függ, hogy ugyanazon ionnak membrán különböző oldalain mekkora koncentrációja van. A legegyszerűbb és leggyakrabban használt membránelektróda az üvegelektróda.
Oldhatatlan sók keverése típusa AgBr, AgCl, AgIés mások némi műanyaggal (gumi, polietilén, polisztirol) vezettek az alkotáshoz ion szelektív elektródák tovább Br-, Cl-, én-, szelektíven adszorbeálja a jelzett ionokat az oldatból a Paneth - Faience - Hahn szabálynak köszönhetően. Mivel a detektálható ionok koncentrációja az elektródán kívül eltér az elektródán belülitől, a membrán felületein eltérőek az egyensúlyok, ami membránpotenciál megjelenéséhez vezet.
A potenciometrikus meghatározások elvégzéséhez egy elektrokémiai cellát állítanak össze egy indikátor referenciaelektródából, amelyet a vizsgált oldatba merítenek és egy potenciométerhez kapcsolnak. A potenciometriában használt elektródák belső ellenállása nagy (500-1000 MOhm), ezért vannak olyan potenciométerek, amelyek összetett elektronikus nagy ellenállású voltmérők. Az elektródarendszer EMF-jének potenciométerekben történő mérésére egy kompenzációs áramkört használnak a cella áramkörében lévő áram csökkentésére.
Leggyakrabban potenciométereket használnak a pH közvetlen mérésére, más ionok koncentrációjának mutatóira. pNa, pK, pNH2, pClés mV. A méréseket megfelelő ion-szelektív elektródákkal végezzük.
A pH mérésére üvegelektródát és referenciaelektródát - ezüst-kloridot - használnak. Az elemzések elvégzése előtt ellenőrizni kell a pH-mérők kalibrálását standard pufferoldatokkal, amelyek rögzítése a készülékhez van rögzítve.
pH-mérők a közvetlen meghatározások mellett pH, pNa, pK, pNH2, pCl mások pedig lehetővé teszik a meghatározandó ion potenciometriás titrálását.
1.3 Potenciometrikus titrálás
A potenciometrikus titrálást olyan esetekben kell elvégezni, amikor a kémiai indikátorok nem használhatók, vagy ha nem áll rendelkezésre megfelelő indikátor.
A potenciometrikus titrálásnál indikátorként a titrált oldatba helyezett potenciométer elektródákat használjuk. Ebben az esetben olyan elektródákat használnak, amelyek érzékenyek a titrált ionokra. A titrálás során az ionkoncentráció változik, amit a potenciométer mérőskáláján rögzítünk. A potenciométer leolvasását pH vagy mV egységben rögzítve ábrázoljuk a titrálószer térfogatától való függését (titrálási görbe), határozzuk meg az ekvivalenciapontot és a titráláshoz felhasznált titrálószer térfogatát. A kapott adatok alapján potenciometrikus titrálási görbét készítünk.
A potenciometrikus titrálási görbe alakja hasonló a titrimetriás analízis titrálási görbéjéhez. A titrálási görbe az ekvivalenciapont meghatározására szolgál, amely a titrálási ugrás közepén található. Ehhez a titrálási görbe egyes szakaszaihoz érintőket rajzolunk, és az ekvivalenciapontot a titrálási ugrás érintőjének közepén határozzuk meg. A legnagyobb változási érték ? pH/?V az ekvivalencia pontján szerzi meg.
Az ekvivalenciapont még pontosabban meghatározható a Gran-féle módszerrel, amelyet a függőség megkonstruálására használnak. ? V/?E a titráló térfogatból. Gran módszerrel a potenciometrikus titrálás anélkül is elvégezhető, hogy az ekvivalenciapontra kerülne.
A titrimetriás elemzés minden esetben potenciometrikus titrálást alkalmaz.
A sav-bázis titráláshoz üvegelektródát és referenciaelektródát használnak. Mivel az üvegelektróda érzékeny a közeg pH-értékének változásaira, titráláskor a közeg pH-értékének változásait a potenciométer rögzíti. A sav-bázis potenciometrikus titrálást sikeresen alkalmazzák gyenge savak és bázisok titrálására (pK?8). Savak keverékének titrálásakor szükséges, hogy pK-értékük 4 egységnél nagyobb különbséggel térjen el, különben a gyengébb sav egy része együtt titrálódik az erős savval, és a titrálási ugrás nem fejeződik ki egyértelműen.
Ez lehetővé teszi a potenciometria használatát kísérleti titrálási görbék felépítésére, a titráláshoz szükséges indikátorok kiválasztására, valamint a savassági és lúgossági állandók meghatározására.
A kicsapásos potenciometrikus titrálásnál indikátorként egy fémből készült elektródát használnak, amely elektródpárt alkot a meghatározandó ionokkal.
Ha komplexometrikus titrálást alkalmazunk: a) fémelektród, amely reverzibilis a meghatározandó fém ionjára; b) platinaelektródát redoxpár jelenlétében az oldatban. Amikor a redoxpár egyik komponensét megköti a titráló, annak koncentrációja megváltozik, ami változást idéz elő az indikátor platinaelektróda potenciáljában. A fémsóhoz adott feleslegben lévő EDTA-oldat vas(III)-só-oldattal történő visszatitrálását is alkalmazzák.
A redox titráláshoz egy referenciaelektródát és egy platina indikátorelektródát használnak, amelyek érzékenyek a redox párokra.
A potenciometrikus titrálás az egyik leggyakrabban használt műszeres analízis módszer egyszerűsége, hozzáférhetősége, szelektivitása és széleskörű lehetőségei miatt.
1.4 Konduktometria. Konduktometrikus titrálás
A konduktometria egy oldat elektromos vezetőképességének mérésén alapul. Ha két elektródát helyezünk egy anyag oldatába, és potenciálkülönbséget alkalmazunk az elektródákon, elektromos áram fog átfolyni az oldaton. Mint minden elektromos vezetőt, a megoldásokat is ellenállás jellemzi R ennek fordított mennyisége pedig az elektromos vezetőképesség L:
Ahol R- ellenállás, Ohm;
Fajlagos ellenállás, Ohm. cm;
S - felülete, cm2.
Ahol L - elektromos vezetőképesség, Ohm-1;
R- ellenállás, Ohm.
A konduktometrikus elemzést konduktométerekkel végzik - olyan eszközökkel, amelyek mérik az oldatok ellenállását. Ellenállás érték szerint R határozza meg az oldatok elektromos vezetőképességét, amely annak fordított értéke L.
Az oldatok koncentrációját közvetlen konduktometriával és konduktometrikus titrálással határozzuk meg. Közvetlen konduktometria az oldat koncentrációjának kalibrációs grafikon segítségével történő meghatározására szolgál. A kalibrációs grafikon elkészítéséhez ismert koncentrációjú oldatok sorozatának elektromos vezetőképességét megmérjük, és összeállítjuk az elektromos vezetőképesség és a koncentráció kalibrációs grafikonját. Ezután megmérjük a vizsgált oldat elektromos vezetőképességét, és a grafikonból meghatározzuk a koncentrációját.
Gyakrabban használt konduktometrikus titrálás. Ebben az esetben a vizsgált oldatot elektródákkal ellátott cellába helyezzük, a cellát mágneses keverőre helyezzük és a megfelelő titrálóval titráljuk. A titrálót egyenlő adagokban adjuk hozzá. A titrálószer minden egyes adagjának hozzáadása után mérje meg az oldat elektromos vezetőképességét, és ábrázolja az elektromos vezetőképesség és a titrálószer térfogata közötti összefüggést. Ha titrálószert adunk hozzá, az oldat elektromos vezetőképessége megváltozik. a titrálási görbe inflexiál.
n az ionok mobilitása függ az elektromos az oldat vezetőképessége: minél nagyobb a mobilitás b ionok, annál nagyobb az elektromos az oldat vezetőképessége.
A konduktometrikus titrálásnak számos előnye van. Zavaros és színes környezetben, kémiai indikátorok hiányában végezhető. A módszer fokozott érzékenységgel rendelkezik, és lehetővé teszi az anyagok híg oldatainak elemzését (10-4 mol/dmі). Az anyagok keverékeit konduktometrikus titrálással elemezzük, mert a különböző ionok mobilitásában mutatkozó különbségek jelentősek, és egymás jelenlétében eltérően titrálhatók.
A konduktometrikus analízis könnyen automatizálható, ha a titrálóoldatot állandó sebességgel bürettából tápláljuk be, és az oldat elektromos vezetőképességének változását rögzítőn rögzítjük.
Ezt a fajta konduktometriát ún chrono- konduktometrikus elemzés.
A sav-bázis titrálásnál konduktometriával meghatározhatók az erős savak, a gyenge savak, a gyenge bázisok sói és az erős savak.
BAN BEN kicsapó konduktometrikustitrálás a titrált oldatok elektromos vezetőképessége először csökken vagy egy bizonyos állandó szinten marad a titrált elektrolit csapadékká kötődése miatt, miután i.e. ha a titrálószer feleslege jelenik meg, az ismét növekszik.
BAN BEN összetettmetrikus konduktometrikus titrálás az oldat elektromos vezetőképességében bekövetkező változások a fémkationok EDTA-val komplexbe kötése miatt következnek be.
Redox konduktometrikustitro- ciója a reagáló ionok koncentrációjának változásán és új ionok megjelenésén alapul az oldatban, ami megváltoztatja az oldat elektromos vezetőképességét.
Az elmúlt években fejlődés történt nagyfrekvenciás konduktometria, amelyben az elektródák nem érintkeznek az oldattal, ami fontos az agresszív közegek és a zárt edényekben lévő oldatok elemzésekor.
Két lehetőséget fejlesztettek ki - egyenesnagyfrekvenciás konduktometria és nagyfrekvenciás titrálás.
Közvetlen nagyfrekvenciás konduktometriát használnak az anyagok, gabona, fa nedvességtartalmának, az oldatok koncentrációjának meghatározására zárt edényekben - ampullákban, valamint az agresszív folyadékok elemzésekor.
A nagyfrekvenciás titrálást speciális titrátorokon végzik - TV-6, TV-6L.
A nagyfrekvenciás konduktometrikus titrálásokat sav-bázis, redox vagy csapadékos titrálásként végezzük olyan esetekben, amikor nem áll rendelkezésre megfelelő indikátor, vagy anyagok keverékének elemzésekor.
1.5 Kulometria. Kulonometrikus titrálás
A coulometriában az anyagokat a mennyiségi elektrokémiai átalakításukra fordított villamos energia mennyiségének mérésével határozzák meg. A coulometriás elemzést elektrolitikus cellában végzik, amelybe a meghatározandó anyag oldatát helyezik. Ha megfelelő potenciált alkalmaznak a cella elektródáin, az anyag elektrokémiai redukciója vagy oxidációja következik be. Az elektrolízis Faraday által felfedezett törvényei szerint az elektródán reagált anyag mennyisége arányos az oldaton áthaladó elektromosság mennyiségével:
Ahol g- a kibocsátott anyag tömege, g;
n- az elektródfolyamat során átvitt elektronok száma;
F- Faraday-szám (F = 96485 C/mol);
én- áramerősség, A;
t- idő, s;
M- a kibocsátott anyag moláris tömege, g/mol.
A coulometriás analízis lehetővé teszi olyan anyagok meghatározását, amelyek elektrokémiai reakció során nem rakódnak le elektródákra, illetve nem jutnak ki a légkörbe.
Létezik közvetlen coulometriás és coulometriás titrálás.. Az elektromos áram mérésére szolgáló módszerek nagy pontossága és érzékenysége egyedülálló, 0,1-0,001%-os pontosságú coulometriás elemzést tesz lehetővé, és 1 10 -8? 1 10 -10 g. Ezért a mikroszennyeződések és az anyagok bomlástermékeinek meghatározására coulometriás analízist alkalmaznak, ami fontos a minőség ellenőrzése során.
Jelezni pl. A coulometriás titrálásnál kémiai és műszeres módszerek alkalmazhatók - indikátorok hozzáadása, színes vegyületek fotometriai vagy spektrofotometriás kimutatása.
Más elemzési módszerekkel ellentétben a coulometria teljesen automatizálható, ami minimalizálja a véletlenszerű meghatározási hibákat. Ezt a funkciót használták az automatikus coulometriás titrátorok létrehozásához – érzékeny műszerek, amelyeket különösen pontos elemzésekhez használnak, amikor más módszerek nem elég érzékenyek. A vízben rosszul oldódó anyagok elemzésekor a coulometriát acetilénfeketéből készült elektródákon lehet elvégezni, amelyek jó adszorbensek és kellő teljességgel eltávolítják az ilyen anyagokat a reakcióközegből. A coulometriás titrálás a műszeres elemzés ígéretes módszere. Széles körben alkalmazható számos speciális analitikai probléma megoldásában - szennyeződések, kis mennyiségű gyógyszerek elemzése, mérgező anyagok, nyomelemek és egyéb vegyületek meghatározása biológiai anyagokban és a környezetben.
KÖVETKEZTETÉS
A munka áttekintést ad a főbb elektrokémiai kutatási módszerekről, részletesen ismertetve azok elvét, alkalmazását, előnyeit és hátrányait.
Az elektrokémiai elemzési módszerek az elektrolízis alkalmazásán alapuló kvantitatív kémiai elemzési módszerek csoportja.
A módszer változatai az elektrogravimetriás analízis (elektroanalízis), a belső elektrolízis, a fémek kontaktcseréje (cementálás), a polarográfiás analízis, a coulometria stb. Az elektrogravimetriás analízis az egyik elektródán felszabaduló anyag mérésén alapul. A módszer nemcsak a réz, nikkel, ólom stb. mennyiségi meghatározását teszi lehetővé, hanem az anyagok keverékeinek elkülönítését is.
Emellett az elektrokémiai elemzési módszerek közé tartoznak az elektromos vezetőképesség mérésén (conduktometria) vagy az elektródpotenciálon (potenciometrián) alapuló módszerek. Egyes elektrokémiai módszereket alkalmaznak a titrálás végpontjának meghatározására (amperometrikus titrálás, konduktometrikus titrálás, potenciometrikus titrálás, coulometriás titrálás).
A HASZNÁLT HIVATKOZÁSOK JEGYZÉKE
1. A modern elektrokémiai elemzés alapjai. Budnikov G.K., Maistrenko V.N., Vyaselev M.R., M., Mir, 2003.
2. J. Plambeck, szerk. S. G. Mayranovsky Elektrokémiai elemzési módszerek. Elméleti és alkalmazási alapok: ford. angolról / Vidannya: Mir, 1985.
3. Damaskin B.B., Petriy O.A., Tsirlina G.A. Elektrokémia - M.: kémia, 2001. 624 p.
4. STO 005-2015. Minőségirányítási rendszer. Oktatási és módszertani tevékenység. Tanfolyami projektek (munkák) és műszaki szakterületek záró minősítő munkáinak nyilvántartása.
Közzétéve az Allbest.ru oldalon
...Hasonló dokumentumok
Az elektrokémiai elemzési módszerek osztályozása, a voltammetria, konduktometria, potenciometria, amperometria, kulometria lényege, alkalmazása a környezetvédelemben. Vegyelemző berendezések és főbb értékesítő cégek jellemzői.
tanfolyami munka, hozzáadva 2010.08.01
Az elektrokémiai módszerek a vizsgált oldatban előforduló elektrokémiai jelenségek elektromos paramétereinek mérésén alapulnak. Az elektrokémiai elemzési módszerek osztályozása. Potenciometrikus, konduktometrikus, coulometriás titrálás.
absztrakt, hozzáadva: 2011.07.01
Az elektrokémiai elemzési módszerek osztályozása. Anyag koncentrációjának potenciometrikus meghatározása oldatban. A konduktometria elve. A konduktometrikus titrálás során fellépő reakciók típusai. Kvantitatív polarográfiai elemzés. Közvetlen coulometria.
tanfolyami munka, hozzáadva 2013.04.04
Az elektroanalitikai módszerek lényege, kísérleti információszerzés képessége a kémiai rendszerek kinetikájáról és termodinamikájáról. A voltammetria, konduktometria, potenciometria, amperometria és coulometria előnyei, hátrányai és alkalmassága.
absztrakt, hozzáadva: 2009.11.20
A potenciometrikus elemzés általános jellemzői. Indikátorelektródák (elektroncserélő és ionszelektív). A potenciometrikus elemzési módszerek típusai. Közvetlen potenciometria és potenciometrikus titrálás. Elektrokémiai áramkörök EMF-jének mérése.
tanfolyami munka, hozzáadva 2012.08.06
Az elektrokémiai elemzési módszerek általános fogalmai, feltételei és osztályozása. Potenciometriai elemzés (potenciometria). Amperometrikus titrálás (potenciometrikus polarizációs titrálás). Kvantitatív polarográfiai elemzés.
absztrakt, hozzáadva: 2012.10.01
Elektrokémiai kutatási módszerek, osztályozásuk és lényegük, előfordulástörténetük. A savkoncentráció meghatározása konduktometrikus titrálással; elektródák potenciálja, galvánelem EMF, réz elektrokémiai megfelelője.
tanfolyami munka, hozzáadva 2014.12.15
A potenciometriai elemzés módszerének tanulmányozása. Kutatási objektumok elemzése, értékelése. A potenciometrikus analízis technikájának tanulmányozása az objektumra vonatkozóan. Hústermékek potenciometriás elemzési módszereinek alkalmazási lehetőségének meghatározása.
tanfolyami munka, hozzáadva 2017.09.16
Alapvető elektrokémiai elemzési módszerek. A potenciometrikus elemzés általános jellemzői. A potenciometrikus elemzési módszerek típusai. Két elektródából álló galvánelem használata. Az indikátorelektróda potenciáljának mérési eljárása.
tanfolyami munka, hozzáadva 2014.08.11
A műszeres elemzési módszerek osztályozása a meghatározandó paraméter és a mérési módszer szerint. A potenciometrikus, amperometrikus, kromatográfiás és fotometriás titrálás lényege. A cink-klorid minőségi és mennyiségi meghatározása.
Az elektrokémiai elemzési módszerek a kvalitatív és kvantitatív elemzési módszerek összessége, amelyek a vizsgált közegben vagy a határfelületen előforduló elektrokémiai jelenségeken alapulnak, és az analit szerkezetének, kémiai összetételének vagy koncentrációjának változásaihoz kapcsolódnak.
Az elektrokémiai elemzési módszerek (ECMA) az elektródákon vagy az elektródák közötti térben végbemenő folyamatokon alapulnak. Előnyük a berendezések és az elemzési módszerek nagy pontossága és viszonylagos egyszerűsége. A nagy pontosságot az ECMA-ban használt nagyon pontos minták határozzák meg. Nagy kényelem, hogy ez a módszer elektromos hatásokat használ, és az a tény, hogy ennek a hatásnak az eredményét (válasz) is megkapjuk elektromos jel formájában. Ez biztosítja az olvasás nagy sebességét és pontosságát, és széles körű automatizálási lehetőségeket nyit meg. Az ECMA-t jó szenzitivitás és szelektivitás jellemzi, esetenként mikroanalízisnek is besorolható, mivel néha 1 ml-nél kevesebb oldat is elegendő az elemzéshez.
Az analitikai jelek típusa szerint a következőkre oszthatók:
1) konduktometria - a vizsgálati oldat elektromos vezetőképességének mérése;
2) potenciometria - az indikátorelektróda árammentes egyensúlyi potenciáljának mérése, amelyre a vizsgált anyag potenciált határoz meg;
3) coulometria - a vizsgált anyag teljes átalakulásához (oxidációjához vagy redukciójához) szükséges villamos energia mennyiségének mérése;
4) voltammetria – az elektródák stacionárius vagy nem stacionárius polarizációs jellemzőinek mérése a vizsgált anyagot érintő reakciókban;
5) elektrogravimetria - az elektrolízis során oldatból felszabaduló anyag tömegének mérése.
27. Potenciometrikus módszer.
potenciometria - az indikátorelektróda árammentes egyensúlyi potenciáljának mérése, amelyre a vizsgált anyag a potenciálást meghatározó.
A) szabvány (referencia elektróda) - állandó potenciállal rendelkezik, független a külső hatásoktól. Feltételek
B) egyedi elektród - potenciálja az anyag koncentrációjától függ.
A potenciál a koncentrációtól függ: E = f(c)
Nerist egyenlet E= E° + lna kat
E° - alapértelmezett. Elektron. Lehetséges (const)
R- Univer. Gáz állandóconst)
T – abszolút hőmérséklet (t)- +273 °
.п – az érintett elektronok száma. Oxidációban/redukcióban Reakciók
. a – aktív koncentráció
Potenciometriai módszer
Ionometrikus potenciometria (kis oldatot adunk a kutatóoldathoz. Standard oldatot (titrán) adagolunk, minden adagolás után mérjük a potenciált. - E)
Egyenértékűségi pont
EСх Vх = l t *Vt
28. Konduktometrikus módszer.
konduktometria - a vizsgálati oldat elektromos vezetőképességének mérése.
Konduktometrikus titrálás
Konduktométer (készülék)
A konduktometriai analízis (conduktometria) az elektrolitoldatok elektromos vezetőképessége (elektromos vezetőképessége) és koncentrációjuk összefüggésének felhasználásán alapul.
Az elektrolit oldatok - a második típusú vezetők - elektromos vezetőképességének megítélése az elektromos ellenállásuk elektrokémiai cellában történő mérése alapján történik, amely egy üvegedény (üveg), amelybe két elektródát forrasztottak, amelyek között a vizsgált elektrolit oldat található. A cellán váltakozó elektromos áram halad át. Az elektródák leggyakrabban fémplatinából készülnek, amelyet az elektródák felületének növelése érdekében szivacsos platinaréteggel vonnak be a platinavegyületek oldatokból történő elektrokémiai leválasztásával (platinizált platinaelektródák).
29.Polarográfia.
A polarográfia kvalitatív és kvantitatív kémiai elemzési módszer, amely a vizsgált oldatból és az abba merített elektródákból álló áramkörben az áram/feszültség görbék megalkotásán alapul, amelyek közül az egyik erősen polarizálható, a másik pedig gyakorlatilag nem polarizálható. Az ilyen görbéket - polarogramokat - polarográfok segítségével kapjuk meg.
A polarográfiás módszert nagy érzékenység jellemzi. Az elemzés elvégzéséhez általában 3-5 ml tesztoldat elegendő. Az automatikus rögzítő polarográf segítségével végzett elemzés mindössze körülbelül 10 percig tart. A polarográfiát a biológiai eredetű tárgyak (például higany-, ólom-, talliumvegyületek) mérgező anyagok tartalmának meghatározására, a vér oxigéntelítettségi fokának meghatározására, a kilélegzett levegő összetételének vizsgálatára használják, és az ipari vállalkozások levegőjében lévő káros anyagok A polarográfiás elemzési módszer rendkívül érzékeny, és nagyon alacsony (akár 0,0001%-os) koncentrációjú anyagok meghatározását teszi lehetővé oldatban.
30. A spektrális elemzési módszerek osztályozása. A spektrum fogalma.
A spektrális elemzés a minőség és a mennyiség meghatározására szolgáló módszerek összessége. Az anyag összetétele és szerkezete (a kutatási objektum különböző típusú sugárzásokkal való kölcsönhatása alapján).
Valamennyi spektroszkópiai módszer a vizsgált anyagot alkotó atomok, molekulák vagy ionok elektromágneses sugárzással történő kölcsönhatásán alapul. Ez a kölcsönhatás a fotonok (kvantumok) abszorpciójában vagy emissziójában nyilvánul meg. A minta elektromágneses sugárzással való kölcsönhatásának természetétől függően a módszerek két csoportját különböztetjük meg:
Emisszió és abszorpció. Attól függően, hogy mely részecskék alkotják az analitikai jelet, megkülönböztetünk atomspektroszkópiai módszereket és molekulaspektroszkópiai módszereket.
Kibocsátás
Az emissziós módszereknél a vizsgált minta gerjesztése következtében fotonokat bocsát ki.
abszorpció
Az abszorpciós módszereknél a külső forrásból származó sugárzás áthalad a mintán, és a kvantumok egy részét szelektíven elnyelik az atomok vagy molekulák
Hatótávolság- egy fizikai mennyiség értékeinek eloszlása (általában energia, frekvencia vagy tömeg). Egy ilyen eloszlás grafikus ábrázolását spektrális diagramnak nevezzük. A spektrum általában az elektromágneses spektrumra utal - az elektromágneses sugárzás frekvenciáinak (vagy a kvantumenergiákkal megegyező) spektrumára.
1.fényvisszaverődés
2. a fénysugár elfordulása (defrakció)
3.fényszórás: nefelometria, turbidimetria
4.fényelnyelés
5 ismételt kibocsátás
A) foszforeszcencia (hosszú ideig tart)
B) fluoreszcencia (nagyon rövid)
A fizikai mennyiségértékek eloszlásának jellege szerint a spektrumok lehetnek diszkrétek (vonalasak), folytonosak (szilárd), valamint diszkrét és folytonos spektrumok kombinációját (szuperpozícióját) is képviselhetik.
A vonalspektrumok példái közé tartoznak a tömegspektrumok és egy atom kötött-kötött elektronátmeneteinek spektrumai; a folytonos spektrumok példái a felhevített szilárd test elektromágneses sugárzásának spektruma és az atomok szabad elektronátmeneteinek spektruma; a kombinált spektrumok példái a csillagok emissziós spektrumai, ahol a kromoszférikus abszorpciós vonalak vagy a legtöbb hangspektrum a fotoszféra folytonos spektrumára helyeződik.
31. Fotometria: a módszer elve, alkalmazása a törvényszéki kutatásban.
Fotometria - spektrális módszer, amely az elektromágneses sugárzás abszorpcióján alapul a látható és közeli ultraibolya tartományban (a módszer a fényelnyelésen alapul)
Molekuláris atom
Spektroszkópia spektroszkópia (elektronanalízisben)
Küvetta - fény áramlik át rajta
l
I (kimeneti fény intenzitása)
I° a beeső fény intenzitása.
A fotometria a fizikai optika és méréstechnika egy része, amely az optikai sugárzás energiajellemzőinek tanulmányozására szolgáló módszereknek szentelt a kibocsátás, a különböző közegekben való terjedés és a testekkel való kölcsönhatás során. A fotometriát infravörös (hullámhossz - 10 -3 ... 7 10 -7 m), látható (7 10 -7 ... 4 10 -7 m) és ultraibolya (4 10 -7 ... 10 -8 m) optikai sugárzás. Amikor az optikai tartomány elektromágneses sugárzása biológiai környezetben terjed, számos fő hatás figyelhető meg: a sugárzás abszorpciója és szóródása a közeg atomjai és molekulái által, a közeg inhomogenitásának részecskék általi szóródása, a sugárzás depolarizációja. Az optikai sugárzás és a közeg kölcsönhatására vonatkozó adatok rögzítésével lehetőség nyílik a vizsgált objektum orvosi és biológiai jellemzőihez kapcsolódó mennyiségi paraméterek meghatározására. A fotometriai mennyiségek mérésére fotométernek nevezett műszereket használnak. Fotometriai értelemben a fény olyan sugárzás, amely az emberi szem számára fényes érzetet kelt. A fotometria mint tudomány alapja az A. Gershun által kidolgozott fénytérelmélet.
A fotometriának két általános módszere van: 1) vizuális fotometria, amely az emberi szem azon képességét használja fel, hogy két összehasonlító mező fényerejét mechanikus vagy optikai eszközökkel kiegyenlítse a fényerő különbségeinek észlelésére; 2) fizikai fotometria, amelyben különböző típusú fényvevőket használnak két fényforrás összehasonlítására - vákuum fotocellák, félvezető fotodiódák stb.
32. Bouguer-Lambert-Beer törvény, felhasználása a mennyiségi elemzésben.
Fizikai törvény, amely meghatározza a párhuzamos monokromatikus fénysugár csillapítását, amikor az elnyelő közegben terjed.
A törvényt a következő képlet fejezi ki:
![]() ,
,

ahol a bejövő sugár intenzitása, az anyagréteg vastagsága, amelyen a fény áthalad, az abszorpciós index (nem tévesztendő össze a dimenzió nélküli abszorpciós indexszel, amely a képlethez kapcsolódik, ahol a hullámhossz) .
Az abszorpciós index az anyag tulajdonságait jellemzi, és az elnyelt fény λ hullámhosszától függ. Ezt a függőséget az anyag abszorpciós spektrumának nevezzük.
A fényelnyelő anyagok nem fényelnyelő oldószerben készült oldatai esetében az abszorpciós indexet a következőképpen írhatjuk fel:
ahol az elnyelő oldott anyag molekulájának λ hullámhosszúságú fénnyel való kölcsönhatását jellemző együttható, az oldott anyag koncentrációja, mol/l.
A nem függő állítást Beer-törvénynek nevezzük (nem tévesztendő össze a Beer-törvénnyel). Ez a törvény azt feltételezi, hogy egy molekula fényelnyelő képességét nem befolyásolják az oldatban lévő azonos anyag többi molekulája. Ettől a törvénytől azonban számos eltérés figyelhető meg, különösen általánosságban.
Ha egy I intenzitású fényáram áthalad egy I vastagságú oldat vagy gáz bizonyos rétegén, akkor a Lambert-Beer törvény szerint az elnyelt fény mennyisége arányos lesz az elnyelő anyag intenzitásával /, c koncentrációjával. fény és a RÉTEG vastagsága) a BMB törvény, amely az anyagra és az azon áthaladó anyagra beeső fény intenzitását az anyag koncentrációjával és az elnyelő réteg vastagságával hozza összefüggésbe. ugyanaz, mint a fénytörés, csak csillapítás az anyagban. Ami bizonyos százalékban elnyeli a fényt. Vagyis a fénykibocsátás fennmaradó része az
33.IR spektroszkópia.
Ez az elemzési módszer egy anyag infravörös abszorpciós spektrumának rögzítésén alapul. Az infravörös tartományban az anyag abszorpciója a molekulákban lévő atomok rezgései miatt következik be. A rezgéseket nyújtásra (amikor az atomok távolsága a rezgés során megváltozik) és vibrációsra (amikor a kötések közötti szögek a rezgés során megváltoznak) osztják. A molekulákban a különböző rezgésállapotok közötti átmeneteket kvantáljuk, aminek köszönhetően az IR tartományban az abszorpció spektrum alakú, ahol minden rezgésnek saját hullámhossza van. Nyilvánvaló, hogy az egyes rezgések hullámhossza attól függ, hogy mely atomok vesznek részt benne, ráadásul kevéssé függ a környezetüktől.
Az IR spektroszkópiai módszer nem elválasztó módszer, azaz bármely anyag vizsgálatakor kiderülhet, hogy valójában több anyag keveréke volt a vizsgált, ami persze nagymértékben torzítja a spektrum megfejtésének eredményét. Nos, nem teljesen korrekt egy anyag egyértelmű azonosításáról beszélni az IR spektroszkópiai módszerrel, mivel a módszer inkább bizonyos funkciós csoportok azonosítását teszi lehetővé, nem pedig azok mennyiségét a vegyületben és az egymással való kommunikációs módszert.
Az IR spektroszkópiai módszerrel polimer anyagok, szálak, festékbevonatok és kábítószerek kutatását végzik (a töltőanyag azonosításakor gyakran használnak szénhidrátokat, köztük poliszacharidokat is). A módszer különösen a kenőanyagok vizsgálatánál nélkülözhetetlen, mivel lehetővé teszi mind a kenőanyag-alap, mind az ehhez az alaphoz esetlegesen hozzáadott adalékok (adalékok) jellegének egyidejű meghatározását.
34. Röntgen-fluoreszcencia analízis.
Az XRF (XRF) az egyik modern spektroszkópiai módszer egy anyag vizsgálatára annak elemi összetételének, azaz elemanalízisének megállapítása érdekében. Különféle elemek elemzésére használható a berilliumtól (Be) az uránig (U). Az XRF módszer a vizsgált anyag röntgensugárzásnak való kitételével kapott spektrum összegyűjtésén és utólagos elemzésén alapul. Besugárzáskor az atom gerjesztett állapotba kerül, ami az elektronok magasabb energiaszintekre való átmenetéből áll. Az atom rendkívül rövid ideig, egy mikroszekundum nagyságrendű gerjesztett állapotban marad, majd visszaáll csendes helyzetbe (alapállapotba). Ebben az esetben a külső héjak elektronjai vagy betöltik a keletkező üresedéseket, és a felesleges energia foton formájában kibocsátásra kerül, vagy az energia a külső héjakról egy másik elektronhoz kerül (Auger elektron)
Ökológia és környezetvédelem: nehézfémek meghatározása talajban, üledékekben, vízben, aeroszolokban stb.
Geológia és ásványtan: talajok, ásványok, kőzetek stb. minőségi és mennyiségi elemzése.
Kohászat és vegyipar: alapanyagok, gyártási folyamat és késztermékek minőségellenőrzése
Festék- és lakkipar: ólomfestékek elemzése
35. Atomemissziós spektroszkópia.
Az atomemissziós spektrális analízis olyan elemelemzési módszerek összessége, amelyek a gázfázisban lévő szabad atomok és ionok emissziós spektrumának vizsgálatán alapulnak. Az emissziós spektrumokat általában a legmegfelelőbb 200 és 1000 nm közötti optikai hullámhossz-tartományban rögzítik.
Az AES (atomi emissziós spektrometria) egy anyag elemi összetételének meghatározására szolgáló módszer a vizsgált minta fényforrásokban gerjesztett atomjainak és ionjainak optikai emissziós spektrumából. Fényforrásként az atomemissziós elemzéshez égő lángot vagy különféle típusú plazmát használnak, beleértve az elektromos szikra- vagy ívplazmát, a lézerszikra plazmát, az induktív csatolású plazmát, az izzító kisülést stb. Az AES a leggyakoribb expressz, nagyon érzékeny módszer az elemek szennyeződéseinek azonosítása és mennyiségi meghatározása gáznemű, folyékony és szilárd anyagokban, beleértve a nagy tisztaságú anyagokat is.
Felhasználási területek:
Kohászat: fémek és ötvözetek összetételének elemzése,
Bányászat: geológiai minták és ásványi nyersanyagok vizsgálata,
Ökológia: víz- és talajelemzés,
Berendezések: motorolajok és egyéb műszaki folyadékok fémszennyeződések elemzése,
Biológiai és orvosi kutatások.
Működési elve.
Az atomemissziós spektrométer működési elve meglehetősen egyszerű. Azon alapul, hogy az egyes elemek atomjai bizonyos hullámhosszúságú fényt - spektrális vonalakat - bocsáthatnak ki, és ezek a hullámhosszak különböző elemeknél eltérőek. Ahhoz, hogy az atomok elkezdjenek fényt kibocsátani, gerjeszteni kell őket - hővel, elektromos kisüléssel, lézerrel vagy más módon. Minél több atom van egy adott elemből a vizsgált mintában, annál fényesebb lesz a megfelelő hullámhosszúságú sugárzás.
Az elemzett elem spektrumvonalának intenzitása az elemzett elem koncentrációján túl számos különböző tényezőtől függ. Emiatt lehetetlen elméletileg kiszámítani az összefüggést a vonal intenzitása és a megfelelő elem koncentrációja között. Éppen ezért az elemzéshez olyan standard mintákra van szükség, amelyek összetételükben hasonlóak a vizsgált mintához. Ezeket a standard mintákat először exponáljuk (égetjük) a készüléken. Ezen égések eredményei alapján minden elemzett elemhez kalibrációs grafikont készítünk, azaz. egy elem spektrális vonala intenzitásának a koncentrációjától való függése. Ezt követően a minták elemzésekor ezeket a kalibrációs grafikonokat használják a mért intenzitások koncentrációkká történő újraszámítására.
A minták előkészítése elemzésre.
Nem szabad megfeledkezni arról, hogy a valóságban több milligramm mintát elemzünk a felületéről. Ezért a helyes eredmények eléréséhez a mintának összetételében és szerkezetében homogénnek kell lennie, és a minta összetételének meg kell egyeznie a vizsgált fém összetételével. A fém öntödében vagy olvasztóban történő elemzésekor ajánlatos speciális öntőformákat használni a minták öntéséhez. Ebben az esetben a minta alakja tetszőleges lehet. Csak az szükséges, hogy a vizsgált minta megfelelő felülettel rendelkezzen és állványba rögzíthető legyen. Speciális adapterek használhatók kis minták, például rudak vagy huzalok elemzésére.
A módszer előnyei:
Érintkezés nélküli,
Lehetőség nagyszámú elem egyidejű mennyiségi meghatározására,
Nagy pontosság,
Alacsony észlelési határok,
Könnyű minta-előkészítés,
Alacsony költségű.
36. Atomabszorpciós spektroszkópia.
módszer a vizsgált anyag elemi összetételének kvantitatív meghatározására atomabszorpciós spektrumok segítségével, amely azon alapul, hogy az atomok képesek-e szelektíven elnyelni az elektromágneses sugárzást bomlás közben. a spektrum részei. A.-a.a. speciális eszközök - abszorpció spektrofotométerek. Az elemzett anyag egy mintáját feloldjuk (általában sók képződésével); az oldatot aeroszol formájában az égő lángjába táplálják. Láng hatására (3000°C) a sómolekulák atomokká disszociálnak, amelyek képesek elnyelni a fényt. Ezután egy fénysugarat vezetnek át az égő lángján, amelynek spektrumában az egyik vagy másik elemnek megfelelő spektrumvonalak találhatók. A vizsgált spektrumvonalakat monokromátor segítségével izoláljuk a teljes sugárzástól, és intenzitásukat rögzítő egység rögzíti. Math. a feldolgozás a következő képlet szerint történik: J = J0 * e-kvI,
ahol J és J0 az áteresztett és beeső fény intenzitása; kv – együttható felszívódás, gyakoriságától függően; I - az elnyelő réteg vastagsága
érzékenyebb, mint az atomerőművek
37. Nefelometria és turbidimetria.
S = log (I°/I) csökkenő intenzitás. Az (I°) oldatban az (I) oldatot kilépő intenzitással el kell osztani =
k-const zavarosság
b – fénysugár úthossza
N az egységenkénti részecskék száma. megoldás
A nefelometriás és turbidimetriás elemzés az oldatban szuszpendált szilárd részecskék fényszórásának jelenségét használja.
A nefelometria egy módszer a kolloid rendszerek diszperziójának és koncentrációjának meghatározására az általuk szórt fény intenzitása alapján. A nefelometria, a méréseket egy speciális készülékben, egy nefelométerben végezzük, melynek működése azon alapul, hogy a vizsgált közeg által szórt fény intenzitását összehasonlítják egy másik közeg által szórt fény intenzitásával, amely etalonként szolgál. A kolloid rendszerek általi fényszórás elméletét, amelyben a részecskék mérete nem haladja meg a beeső fény félhullámhosszát, J. Rayleigh angol fizikus dolgozta ki 1871-ben. Rayleigh törvénye szerint az I fény intenzitása a fényre merőleges irányba szóródik. a beeső sugarat a következő képlet fejezi ki: I = QNvlk - ahol q a beeső fény intenzitása, N az egységnyi térfogatra jutó részecskék teljes száma vagy részleges koncentrációja, v egy részecske térfogata, \ a beeső fény hullámhossza a beeső fény, k a kolloid részecskék és az őket körülvevő diszperziós közeg törésmutatóitól, a fényforrástól való távolságtól, valamint az elfogadott mértékegységektől függő állandó
A turbidimetria a zavaros közegek elemzésére szolgáló módszer, amely az általuk elnyelt fény intenzitásának mérésén alapul. A turbidimetriás méréseket áteresztő fényben vizuális turbidiméterekkel vagy fotoelektromos koloriméterekkel végzik. A mérési technika hasonló a kolorimetrikushoz, és a Bouguer-Lambert törvény zavaros közegekre való alkalmazhatóságán alapul, amely szuszpenziók esetén csak nagyon vékony rétegekre vagy jelentős hígításokra érvényes. A turbidimetria megköveteli a diszpergált fázis kialakulásának körülményeinek gondos betartását, hasonlóan a nefelometriánál megfigyelt feltételekhez. A turbidimetria jelentős előrelépése a fotoelektromos koloriméterekkel végzett turbidimetriás csúcsturbiditás-titrálás alkalmazása. A turbidimetriát sikeresen alkalmazzák szulfátok, foszfátok, kloridok, cianidok, ólom, cink stb. analitikai meghatározására.
A nefelometrikus és turbidimetriás módszerek fő előnye a nagy érzékenységük, ami különösen értékes olyan elemek vagy ionok esetében, amelyeknél nincs színreakció. A gyakorlatban például széles körben alkalmazzák a klorid és szulfát nefelometriás meghatározását természetes vizekben és hasonló tárgyakban. A pontosság tekintetében a turbidimetria és a nefelometria alulmúlja a fotometriás módszereket, ami főként az azonos szemcseméretű szuszpenziók előállításának nehézségeiből, az időbeni stabilitásból stb. tudható be. A fotometriai meghatározás szokásos viszonylag kis hibái mellett a hibákhoz is társulnak a kémiai analitikai módszerek elégtelen reprodukálhatósága mellett a szuszpenziók tulajdonságai is hozzáadódnak.
Nefelometriát és turbidimetriát alkalmaznak például a SO4 meghatározására BaSO4 szuszpenzió formájában, Cl- AgCl szuszpenzió formájában, S2- meghatározására CuS szuszpenzió formájában az aljáról. a kimutatható tartalom határa ~ 0,1 μg/ml. A kísérleti analízis körülményeinek standardizálásához szigorúan ellenőrizni kell a hőmérsékletet, a szuszpenzió térfogatát, a reagensek koncentrációját, a keverési sebességet és a mérési időt. A csapadéknak gyorsan kell lezajlania, a kicsapódott részecskéknek kis méretűeknek és alacsony pH-júaknak kell lenniük. A nagy részecskék koagulációjának megakadályozása érdekében gyakran adnak az oldathoz például stabilizátort. zselatin, glicerin.
38. Kromatográfia: eredettörténet, módszer elve, bírósági alkalmazás. Kutatás.
A kromatográfia egy dinamikus szorpciós módszer anyagkeverékek szétválasztására és elemzésére, valamint az anyagok fizikai-kémiai tulajdonságainak tanulmányozására. Az anyagok két fázis – álló (szilárd fázis vagy közömbös hordozón kötött folyadék) és mobil (gáz- vagy folyadékfázis, eluens) – közötti eloszlásán alapul. A módszer nevéhez fűződnek az első kromatográfiás kísérletek, amelyek során a módszer kidolgozója, Mikhail Tsvet élénk színű növényi pigmenteket választott le.
A kromatográfiás módszert először Mihail Szemenovics Cvet orosz botanikus alkalmazta 1900-ban. Kalcium-karbonáttal töltött oszlopot használt a növényi pigmentek elkülönítésére. Az első jelentést a kromatográfiás módszer fejlesztéséről Tsvet készítette 1901. december 30-án, Természetkutatók és Orvosok XI. Kongresszusa Szentpéterváron. Az első nyomtatott kromatográfiás munka 1903-ban jelent meg a folyóiratban A Varsói Természetkutatók Társaságának közleménye. Első ciklus kromatográfia a Color két nyomtatott munkájában jelent meg 1906-ban, egy német folyóiratban Berichte der Deutschen Botanischen Gesellschaft. 1907-ben Tsvet bemutatja módszerét Német Botanikai Társaság.
1910-1930-ban a módszer méltatlanul feledésbe merült és gyakorlatilag nem is fejlődött.
1931-ben R. Kuhn, A. Winterstein és E. Lederer kromatográfiával α és β frakciókat izoláltak kristályos formában a nyers karotinból, ezzel demonstrálva a módszer preparatív értékét.
1941-ben A. J. P. Martin és R. L. M. Singh kifejlesztett egy új típusú kromatográfiát, amely a szétválasztott anyagok eloszlási együtthatóinak különbségén alapult két nem elegyedő folyadék között. A módszer az úgynevezett " megoszlási kromatográfia».
1947-ben T. B. Gapon, E. N. Gapon és F. M. Shemyakin kidolgozta az „ioncserélő kromatográfia” módszerét.
1952-ben J. Martin és R. Singh kémiai Nobel-díjat kapott a megoszlási kromatográfia módszerének megalkotásáért.
A 20. század közepétől napjainkig a kromatográfia intenzíven fejlődött, és az egyik legszélesebb körben használt analitikai módszerré vált.
Besorolás: gáz, folyékony
A kromatográfia alapjai folyamat. A kromatográfiás vizsgálat elvégzéséhez anyagok szétválasztása vagy fizikai-kémiai tulajdonságaik meghatározása. jellemzőit általában speciális. eszközök - kromatográfok. Alapvető kromatográfiás egységek - kromatográfiás. oszlop, detektor és mintainjektáló készülék. A szorbenst tartalmazó oszlop azt a funkciót látja el, hogy a vizsgált keveréket alkotó komponensekre, a detektor pedig a mennyiségek szétválasztását látja el. definíciók. Az oszlop kimeneténél elhelyezett detektor automatikusan folyamatosan meghatározza a leválasztott vegyületek koncentrációját. a mozgófázis áramlásában Miután az elemzett keveréket a mozgófázis áramlásával az oszlopba vezettük, az összes anyag zónái a kromatográfia elején találhatók. oszlopok (1. ábra). A mozgófázis áramlásának hatására a keverék komponensei bomlás közben elkezdenek mozogni az oszlop mentén. sebességek, amelyek értéke fordítottan arányos a kromatografált komponensek K eloszlási együtthatóival. A jól felszívódó anyagok, amelyek eloszlási állandó értékei nagyok, lassabban mozognak a szorbens réteg mentén az oszlop mentén, mint a rosszul felszívódó anyagok. Ezért az A komponens hagyja el leggyorsabban az oszlopot, majd a B komponens, és az utolsó, amelyik elhagyja az oszlopot, a C komponens (KA<К Б <К В). Сигнал детектора, величина к-рого пропорциональна концентрации определяемого в-ва в потоке элюента, автоматически непрерывно записывается и регистрируется (напр., на диаграммной ленте). Полученная хроматограмма отражает расположение хроматографич. зон на слое сорбента или в потоке подвижной фазы во времени.

Rizs. 1. Három komponens (A, B és C) keverékének szétválasztása K kromatográfiás oszlopon D detektorral: a - az elválasztott komponensek kromatográfiás zónáinak helyzete az oszlopban meghatározott időközönként; b - kromatogram (C - jel, t - idő) .
Lapos réteg kromatográfiával Elválasztáskor egy papírlapot vagy egy szorbens réteggel ellátott lemezt helyeznek el a vizsgált anyag mintáival kromatográfiás eljárásban. kamera. Az elválasztás után a komponenseket bármilyen alkalmas módszerrel meghatározzuk.
39. A kromatográfiás módszerek osztályozása.
A kromatográfia az anyagok elválasztásának és elemzésének módszere, amely az analit eloszlásán alapul. A különbség a 2 fázis között: mobil és álló
Az elválasztandó anyagok keverékének oldatát adszorbenssel megtöltött üvegcsövön (adszorpciós oszlopon) vezetjük át. Ennek eredményeként a keverék komponensei az adszorbens oszlop különböző magasságaiban maradnak meg külön zónák (rétegek) formájában. A cucc jobb, mint az adsorbir. Az oszlop tetején vannak, és rosszabbul adszorbeálódnak az oszlop alján. Az adszorbeálhatatlan anyagok megállás nélkül áthaladnak az oszlopon, és a szűrőben gyűlnek össze.
Besorolások:
1. A fázisok összesítési állapota szerint.
1) Mozgatható
A) gáz (inert gázok: hélium, argon, azon)
B) folyadék
2. a megvalósítás módja szerint
1) síkon (síkban); vékonyrétegű papír
2) oszlopos
A) töltve (szorbenssel töltött töltött oszlop)
B) kapilláris (vékony üveg/kvarc kapilláris, amelynek belső felületén állófázis van felhordva)
Def. Tételek kis mennyiségben.
Az illékony anyagokat elkülönítjük.
40. Kromatogram. A kromatográfiás csúcs alapvető paraméterei.
A kromatogram az oszlop kimeneténél lévő komponensek koncentrációjának időbeli függőségének rögzítésének eredménye.
H S
A kromatogram minden csúcsát kettő jellemez fő paraméterei
1. Retenciós idő ( t R) az elemzett minta bevezetésének pillanatától a kromatográfiás csúcs maximumának rögzítéséig eltelt idő. Ez az anyag természetétől függ, és minőségi jellemző.
2. Magasság ( h) vagy terület ( S) csúcs
S = ½ ω × h. (4)
A csúcs magassága és területe az anyag mennyiségétől függ, és mennyiségi jellemzők.
A retenciós idő két összetevőből áll - az anyagok tartózkodási idejéből a mozgófázisban ( t m) és a tartózkodási idő az állófázisban ( t s):
Az elemzett keverék ismeretlen komponenseinek csúcsainak azonosítása összehasonlítással (összehasonlítás) történik. közvetlenül a kromatogramból meghatározott értékek, az ismert vegyületek megfelelő táblázatos adataival. A kromatográfiás azonosítás során csak a negatív megbízható. válasz; például az i csúcs nem A elem, ha az i csúcs és az A retenciós ideje nem esik egybe. Az i csúcs és az A elem retenciós idejének egybeesése szükséges, de nem elégséges feltétele annak a következtetésnek, hogy az i csúcs az A.
A gyakorlati munkában a kromatogramok kvantitatív értelmezésének egyik vagy másik paraméterének megválasztását több tényező együttes hatása határozza meg: a számítás sebessége és egyszerűsége, a kromatográfiás csúcs alakja (széles, keskeny) és aszimmetria foka, az alkalmazott oszlop hatékonysága, a keverék komponenseinek szétválasztásának teljessége, a szükséges automatizált eszközök (integrátorok, kromatográfiás adatok feldolgozására szolgáló számítógépes rendszerek) megléte.
A kromatográfiás csúcs meghatározott paraméterét a kezelõ manuálisan méri a kromatogramon az elemzett keverék komponenseinek elválasztási ciklusának végén.
A kromatográfiás csúcs meghatározott paraméterét digitális voltmérők, integrátorok vagy speciális számítógépek segítségével automatikusan mérik, egyidejűleg az elemzett keverék komponenseinek szétválasztásával az oszlopban és a kromatogram rögzítésével.
Mivel a kromatogramok dekódolásának technikája a kérdéses vegyület és a standard vegyület kromatográfiás csúcsai paramétereinek mérésén alapul, a kromatográfiás körülményeknek biztosítaniuk kell azok lehető legteljesebb elválasztását; az eredeti minta összes többi komponense az elfogadott elemzési feltételek mellett nem válnak el egymástól, vagy egyáltalán nem jelennek meg a kromatogramon (ez a belső standard módszer előnye a belső normalizálási módszerrel szemben)
41.Kvalitatív kromatográfiás elemzés.
Megfelelő oszlophosszúsággal bármely keverék komponenseinek teljes szétválasztása elérhető. És miután az elválasztott komponenseket külön frakciókba (eluátumokba) eluáltuk, határozza meg a keverék komponenseinek számát (az eluátumok számának felel meg), határozza meg azok minőségi összetételét, határozza meg mindegyik mennyiségét megfelelő kvantitatív elemzési módszerekkel. .
Kvalitatív kromatográfiás elemzés, i.e. az anyag kromatogramja szerinti azonosítása elvégezhető a kromatográfiás jellemzők, leggyakrabban a visszatartott térfogat (azaz az oszlopon áthaladó mozgófázis térfogata a keverék bemenetétől a komponens megjelenéséig a kimeneten) összehasonlításával. oszlop), bizonyos feltételek mellett megtalálható az elemzett anyagkeverékek összetevőinél és a szabványnál.
42.Kvantitatív kromatográfiás elemzés.
A kvantitatív kromatográfiás analízist általában kromatográfon végzik. A módszer a kromatográfiás csúcs különböző paramétereinek mérésén alapul, a kromatográfiás anyagok koncentrációjától függően - magasság, szélesség, terület és visszatartott térfogat vagy a visszatartott térfogat és csúcsmagasság szorzata.
A kvantitatív gázkromatográfiában az abszolút kalibrálás és a belső normalizálás, illetve normalizálás módszereit alkalmazzák. A belső standard módszert is alkalmazzák. Abszolút kalibrációval kísérletileg meghatározzák a csúcsmagasság vagy -terület függését az anyag koncentrációjától, és kalibrációs grafikonokat készítenek, vagy kiszámítják a megfelelő együtthatókat. Ezután meghatározzuk az elemzett keverék csúcsainak azonos jellemzőit, és a kalibrációs grafikonon meghatározzuk az analit koncentrációját. Ez az egyszerű és pontos módszer a szennyeződésnyomok meghatározására a fő módszer.
A belső normalizálási módszer alkalmazásakor a csúcsparaméterek összegét, például az összes csúcs magasságának összegét vagy területük összegét 100%-nak vesszük. Ekkor az egyes csúcsok magasságának és a magasságok összegének aránya, vagy egy csúcs területének és a területek összegének aránya 100-zal szorozva jellemzi a komponens tömeghányadát (%) a keverék. Ennél a megközelítésnél szükséges, hogy a mért paraméter értékének koncentrációtól való függése a keverék minden komponensénél azonos legyen.
43.Síkkromatográfia. Vékonyréteg-kromatográfia használata tintaelemzéshez.
A cellulóz vékonyréteg-kromatográfiában való felhasználásának első formája a papírkromatográfia volt. A rendelkezésre álló TLC és nagy áteresztőképességű vékonyréteg-kromatográfiás lemezek lehetővé teszik poláros anyagok keverékeinek szétválasztását, legalább háromkomponensű víz, nem elegyedő szerves oldószer és egy olyan vízben oldódó oldószer keverékének felhasználásával, amely elősegíti az egyik fázis kialakulását.